Neurofibromatosis type 1 er en genetisk sykdom som misdannelser i sentralnervesystemet og huden er karakteristiske for. Med rundt ett av 3000 nyfødte er nevrofibromatose type 1 en av de vanligste genetiske sykdommene.
Hva er nevrofibromatose type 1?
© peshkova - lager.adobe.com
Som Neurofibromatosis type 1 (også Recklinghausen sykdom) er en genetisk fakomatose med misdannelser i huden og sentralnervesystemet.
Neurofibromatosis manifesterer seg på grunnlag av pigmentanomalier som allerede forekommer i spedbarnsalderen, for eksempel flekker med en kaffebrun farge (café-au-lait flekker eller melkekaffe flekker) og axillære og inguinal lentigines (fregne-lignende pigmentering i armhulene og lysken).
Et annet nøkkelsymptom på nevrofibromatose type 1 er nevrofibromene som finnes over hele kroppen til personen som er rammet. Nevrofibromer er godartede (godartede) svulster som oppstår i de fleste tilfeller i barndommen og kan føre til smerter og nevrologiske mangler som parestesi hvis, i tillegg til huden, påvirkes indre organer, spesielt ryggmargsnervene og hjernen.
I tillegg i nevrofibromatose av denne typen er benavvik som skoliose (krumning i ryggraden), irsk amartom (Lisch knuter på iris og fremre øye), og vansker med å lære og konsentrere seg.
fører til
EN Neurofibromatosis type 1 skyldes mutative genendringer, hvorved Recklinghausens sykdom har en mutasjon av det såkalte NF-1-genet (neurofibromatosis 1-genet) på det 17. kromosomet, noe som fører til ukontrollert celleproliferasjon (celleproduksjon og vekst).
Mutasjonen som er ansvarlig for manifestasjonen av sykdommen, blir enten gitt videre til barnet fra en av foreldrene via den autosomale dominante arven eller utvikler seg som et resultat av nye mutasjoner som spontane endringer i genomet til den berørte personen (i rundt 50 prosent av tilfellene).
Det er foreløpig ikke kjent hvilke faktorer som utløser disse mutative prosessene. Det antas at størrelsen på det berørte kromosomet, som øker sannsynligheten for mutasjoner, spiller en avgjørende rolle.
Symptomer, plager og tegn
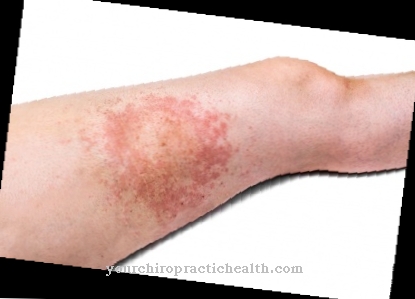
Nevrofibromatose av type 1 er primært preget av forskjellige, for det meste godartede hudforandringer. Dette inkluderer overdreven pigmentering av huden, som allerede kan forekomme hos babyer og nyfødte. Formen deres er ofte oval, mens fargen vagt minner om melkekaffe. Derfor er dette også kjent som café-au-lait flekker i medisin.
Fregne-lignende flekker vises noen ganger i armhulene, lysken eller til og med slimhinnen i munnen. Når sykdommen utvikler seg, vokser godartede knuter på overflaten av huden, noen av dem betydelige i størrelse, flere centimeter fra tiårsalderen. Disse nevrofibromene forekommer også spontant i svangerskapet.
Som regel forårsaker ikke svulstene og hudlesjonene smerter eller annet ubehag.Derfor, bortsett fra den estetiske svekkelsen, forventer de berørte et sunt og lidelsesfritt livsløp. Veksten av neurofibromer er ikke begrenset til hudoverflaten. De kan også utvikles på iris eller i kroppen. Godartede svulster på synsnerven hindrer utsikten (optisk gliom).
Nervefibersvulster og buede bein (skoliose) setter også en belastning på ytelsen. Læringsproblemer og tap av konsentrasjon er resultatet. I ekstreme tilfeller kan vekstene også forårsake epileptiske anfall. I mildere form kan synsforstyrrelser, symptomer på lammelse eller en generell tendens til unormale sensasjoner observeres. Nevrofibromer av type 1 utvikler seg sjelden til ondartede svulster og forårsaker følgelig smerte for de som lider avhengig av hvor de befinner seg.
Diagnose og kurs
EN Neurofibromatosis type 1 diagnostiseres basert på symptomene som er karakteristiske for sykdommen som café-au-lait flekker, neurofibromer, flere lentiginer, Lisch knuter. Den underliggende mutasjonen på kromosom 17 (NF-1 gen) kan påvises i løpet av en DNA-analyse.
En familiær ansamling av sykdommen kan også indikere nevrofibromatose. Hvis den spesifikke mutasjonen i familien er kjent, kan dette bestemmes prenatalt ved hjelp av en korionisk villusprøvetaking eller en fostervannsprøve. Avbildningsmetoder som røntgenstråler (skoliose) eller elektroencefalografi (nedsatt hjerne) brukes for å bestemme om indre organer er involvert.
Selv om kursene kan variere veldig selv i familien, har nevrofibromatose et mildt forløp i de fleste tilfeller (rundt 60 prosent). Hvis ubehandlet, kan nevrofibromatose føre til uttalte svikt som svulster i synsnerven eller hjernen.
komplikasjoner
Hvorvidt det er komplikasjoner i nevrofibromatose type 1, varierer veldig. Noen sykdomstilfeller tar et mildt forløp, mens andre lider kan få alvorlige konsekvenser. Av denne grunn er regelmessige kontroller ekstremt viktig fordi legen kan identifisere eventuelle effekter som krever behandling på et tidlig tidspunkt.
De såkalte delvise prestasjonsforstyrrelsene er blant de vanligste følgene av nevrofibromatose type 1. Dette fører til lærevansker hos barn som faktisk har normal intelligens. Omtrent halvparten av alle syke barn er rammet av læringsproblemer. Det er ikke uvanlig at atferdsforstyrrelser, oppmerksomhetsmangel eller hyperaktivitet oppstår parallelt. Imidlertid er det ingen forverring i læringsvansker. De berørte barna skal få støtte før de begynner på skolen.
En annen konsekvens av nevrofibromatose type 1 er utseendet av nevrofibromer under huden. Hos noen pasienter vises de i barndommen, i andre bare i puberteten. Eksisterende neurofibromer kan vokse i puberteten. Nevrologiske forstyrrelser eller smerter er mulig, avhengig av hvilken del av kroppen svulstene vises.
Krumninger i ryggraden eller skoliose er også utbredt blant pasienter med nevrofibromatose. De er mer vanlig i nevrofibromatose enn hos friske mennesker. Alvorlige komplikasjoner er hjernesvulster eller svulster i synsnerven som optiske gliomer. I tillegg er epileptiske anfall mulig.
Når bør du gå til legen?
Foreldre som merker de typiske hudforandringene hos barnet, bør kontakte barnelegen umiddelbart. Medisinske råd er nødvendig senest når det dannes vekster eller knuter på huden. Legen kan diagnostisere nevrofibromatose av type 1 og starte behandling umiddelbart.
Hvis hudlesjonene blir smittet, må barnet få legehjelp samme dag. Hvis det er feber og andre ledsagende symptomer, er det nødvendig å foreta en undersøkelse på sykehuset på sykehuset. I tillegg til familielegen eller barnelegen, kan nevrofibromatose type 1 sees av en hudlege, nevrolog, nevrokirurg eller annen internist.
Selve behandlingen foregår vanligvis i en spesialistklinikk for hudsykdommer. For å sikre optimal pleie, kan tverrfaglige råd være nyttige. Siden sykdommen også kan representere en betydelig belastning for foreldrene, er terapeutiske råd alltid nyttige. Etter avsluttet behandling må barnet undersøkes av lege minst en gang i året. Legen kan avklare om nye svulster har utviklet seg og undersøke de indre organene så vel som øynene og ørene for ytterligere skade.
Behandling og terapi
Fordi den underliggende årsaken er en Neurofibromatosis type 1 er genetisk eller mutativ, kan det ikke behandles kausalt. De terapeutiske tiltakene er rettet mot å redusere de spesifikke symptomene og forhindre potensielle sekundære symptomer.
Nevrofibromer som forårsaker svekket smerte og / eller har risiko for degenerasjon, blir vanligvis fjernet (mikro) kirurgisk som en del av en kirurgisk prosedyre. Svulstene kan fjernes med en skalpell, en laser eller som en del av en elektrokauteri. Mens nevrofibromer lokalisert over hudnivået blir kuttet ut med en skalpell, kan kutane svulster eller svulster lokalisert på hudnivået fjernes med laseren eller ved elektrokauteri.
Sistnevnte gjør det mulig å akselerere hemostasen på samme tid på grunn av effekten av varme, noe som er en avgjørende fordel, spesielt når det gjelder neurofibromer som er rike på blodkar. I prinsippet bør imidlertid en omfattende risiko-fordel-analyse utføres i forkant av inngrepet, siden hver kirurgisk inngrep kan føre til lammelse som et resultat av en funksjonssvikt i de proksimale nervene.
I tillegg kan svulster i området til sentralnervesystemet lokaliseres ugunstig, slik at selv sunne vevsstrukturer kan bli skadet av et kirurgisk inngrep. Radioterapeutiske tiltak for degenererte svulster bør også undersøkes nøye på grunn av økt risiko for ytterligere degenerasjon.
Sist, men ikke minst, må symptomer på nevrofibromatose av type 1 som epilepsi (karbamazepin, klonazepam), dårlig konsentrasjon (endring i kosthold, konsentrasjonsøvelser) eller skoliose (fysioterapi, korsett, kirurgi) behandles.
Outlook og prognose
Personer med nevrofibromatose av type 1 lider av en uhelbredelig sykdom. En genetisk defekt ble funnet å være årsaken til helseproblemet. Siden inngrep og endringer i menneskets genetikk ikke er tillatt på grunn av lovkrav, kan ingen helbredelse finne sted. Dette representerer en betydelig belastning for de berørte og deres pårørende. I medisinsk behandling konsentrerer legene seg om å lindre de eksisterende og individuelt uttalte symptomene. I tillegg er et mål med behandlingen å kontrollere progresjonen av sykdommen.
Takket være medisinske fremskritt er det nå forskjellige behandlingsmetoder som fører til betydelig symptomlindring. Regelmessig medisinsk overvåking brukes også som et forebyggende tiltak for eventuelle komplikasjoner. I mange tilfeller er det forventet svekkelse av sensorisk persepsjon og motoriske lidelser. Unnlatelse av å søke medisinsk behandling øker risikoen for alvorlige følgere. I tillegg er den generelle livskvaliteten betydelig svekket.
I mange tilfeller trenger de som rammes intensiv daglig pleie, da det ikke er mulig å takle hverdagen for dem alene. Pasienter må vanligvis gjennomgå gjentatte kirurgiske inngrep i løpet av livet. Vevsvekster bør fjernes i disse slik at symptomer på svikt kan reduseres eller eksisterende klager kan komme tilbake.
forebygging
Der Neurofibromatosis type 1 er en genetisk eller mutativ sykdom, kan den ikke direkte forhindres. For å forhindre komplikasjoner, bør imidlertid regelmessige kontroller utføres slik at terapeutiske tiltak kan settes i gang i god tid, spesielt hvis det er neurofibromer som kan degenerere til lymfomer eller nevrofibrose.
ettervern
Når det gjelder nevrofibromatose type 1, er det i de fleste tilfeller bare svært få og bare svært begrensede tiltak og alternativer for oppfølging. Av denne grunn må de som er rammet av denne sykdommen konsultere lege veldig tidlig for å unngå andre komplikasjoner eller klager. Som regel kan ikke helbredelse skje.
På grunn av den genetiske sykdommen er det heller ingen fullstendig kur. Derfor, hvis personen det gjelder ønsker å få barn, bør han absolutt foreta en genetisk undersøkelse og råd for å forhindre gjentakelse av nevrofibromatose type 1 hos barna. Under behandlingen er de berørte for det meste avhengige av hjelp og støtte fra sin egen familie.
Det er ikke uvanlig at psykologisk støtte er nødvendig for å forhindre depresjon og andre psykologiske opprør. I mange tilfeller trenger barn med nevrofibromatose type 1 også intensiv støtte på skolen slik at det ikke oppstår komplikasjoner i voksen alder. Fysioterapi og fysioterapi er også veldig viktig, og mange av øvelsene fra disse behandlingen kan også utføres i ditt eget hjem.
Du kan gjøre det selv
Pigmentforandringene som ofte forekommer i forbindelse med nevrofibromatose type 1, kan behandles kosmetisk av pasienten selv, for eksempel med passende kremer. Deler av kroppen som er utsatt for endringer i pigmentering, bør unngås fra intenst sollys.
De mentale funksjonsnedsettelsene som noen ganger er assosiert med nevrofibromatose type 1 (f.eks. Delvise prestasjonsforstyrrelser, atferdsproblemer eller motoriske restriksjoner) kan utføres uavhengig med passende terapier - hvis instruert av en terapeut. I tillegg til konvensjonell medisinsk behandling er selvhjelpsgrupper den viktigste pilaren. Selvhjelpsgrupper tilbyr pasienten et bredt spekter av tilbud, for eksempel å gi terapi, forklare sykdomsforløpet og behandlingsformene, og tilby råd og støtte, for eksempel om genetisk undersøkelse av et foster i svangerskapet. Det er selvhjelpsgrupper i hver føderal stat.
Nevrofibromatose av type 1 utvikler seg annerledes hos hver pasient; Å leve med usikkerhet - når vil nevrofibromatose type 1 bryte ut, hva som egentlig vil skje da - er uunngåelig en del av pasientens liv. Det anbefales, muligens med støtte fra en passende terapeut, å finne måter ut av denne utilfredsstillende situasjonen for å kunne integrere usikkerheten i hverdagen.
.jpg)
.jpg)
.jpg)
.jpg)







.jpg)
.jpg)
.jpg)
.jpg)