De Allan-Herndon-Dudley-syndrom er en mutasjon i SLC16A2-genet som endrer skjoldbruskkjertelhormonetransportøren MCT8 og forårsaker nedsatt jodtyroninopptak i muskelvev og sentralnervesystemet. På grunn av mutasjonen lider de berørte av muskelsvakhet samt forsinkelser i mobil og mental utvikling. AHDS er uhelbredelig og har hittil bare blitt behandlet med administrering av triiodothyroacetate.
Hva er Allan - Herndon - Dudley syndrom?

Forsinkelser i den fysiske, mentale eller emosjonelle utviklingen hos ungdom og barn oppsummeres som forsinkelser eller utviklingshemning. Forsinkelser i utviklingen kan ha forskjellige årsaker. For eksempel kan utløseren for den forsinkede utviklingen ligge i sentralnervesystemet.
Dette er for eksempel tilfellet med Allan - Herndon - Dudley Syndrome (AHDS). I tillegg til alvorlig psykisk utviklingshemning, er syndromet preget av forstyrrelser i motorisk utvikling. Den første beskrivelsen av syndromet går tilbake til 1944. Det kliniske bildet du beskriver er en arvelig sykdom, dvs. en genetisk lidelse.
Sykdommen rammer mannlige spedbarn i de fleste av alle tilfeller dokumentert til dags dato. Utviklingsforstyrrelsene og konsekvensene av disse er i nesten alle tilfeller tydelig synlige fra fødselen. AHDS er en ekstremt sjelden sykdom.Av denne grunn har tilstanden til forskning på Allan - Herndon - Dudley syndrom hittil vært ganske dårlig.
fører til
AHDS er en arvelig genetisk lidelse forårsaket av en mutasjon i SLC16A2-genet. Dette er det kodende genet for den såkalte skjoldbruskhormonetransportøren MCT8. Denne transportøren formidler opptaket av jodtyroniner i muskel- og nervevev.
På grunn av mutasjonen oppstår forstyrrelser i opptaket av skjoldbruskhormoner, som forstyrrer sentralnervesystemet og dermed svekker utviklingen av nervesystemets celler. Muskelvev og hjerne blir fattige på grunn av den mutasjonsrelaterte dysreguleringen av det aktive skjoldbruskhormonet som de faktisk er avhengige av.
Syndromet videreføres som en X-koblet recessiv arv. Kvinner kan arve sykdommen, men på grunn av deres doble X-kromosomstruktur blir de sjelden syke. Syke menn klarer ikke å formere seg.
Selv om mutasjonen er genetisk, spiller i tillegg til denne interne faktoren sannsynligvis eksterne faktorer en rolle i sykdommens begynnelse. På grunn av sjeldenhetene og det begrensede forskningsbasen, har rollen til disse eksterne faktorene ennå ikke blitt endelig avklart.
Du finner medisinene dine her
➔ Medisiner for muskelsvakhetSymptomer, plager og tegn
Allan-Herndon-Dudley syndrom er en medfødt sykdom som vanligvis manifesterer seg hos småbarn eller babyer. De berørte lider av mer eller mindre alvorlig muskelsvakhet. Barnas muskelvev er merkbart underutviklet. Muskels svakhet er snart ledsaget av leddeformiteter.
Kontraksjoner er også hyppige ledsagende symptomer. Barnas mobilitet svekkes i økende grad av kontrakturer og deformiteter. Av denne grunn virker de berørte ofte unaturlig statiske eller til og med ubevegelige. På grunn av det mutasjonsrelaterte underforsyningen av skjoldbruskhormoner lider de som rammes ofte av muskelkramper eller foretar ufrivillige bevegelser med armer og ben.
Ofte kan ikke de berørte bevege seg uavhengig. I de fleste tilfeller er motoriske svekkelser forbundet med alvorlige psykiske lidelser. For eksempel er flertallet av pasientene ikke i stand til å snakke. I enkelttilfeller kan AHDS karakteriseres av mange andre symptomer innen mental og fysisk utvikling.
Diagnose og kurs
Legen mistenker først AHDS i anamnese. I laboratorietester indikerer et økt T3-nivå med normale FT4- og TSH-nivåer Allan - Herndon - Dudley syndrom. Avbildning av sentralnervesystemet er vanligvis en del av diagnosen.
Muskelsvakheter på grunn av motoriske nevronsykdommer kan utelukkes fra differensialdiagnosen. Prognosen for pasienter med Allan-Herndon-Dudley syndrom er relativt dårlig. Så langt er sykdommen uhelbredelig. Studier har antydet at tidspunktet for diagnosen kan spille en kritisk rolle i pasientprognosen.
komplikasjoner
Som alle kromosonalt arvelige lidelser, kan ikke Allan - Herndon - Dudley syndrom behandles kurativt. Den vanligste bivirkningen av Allan - Herndon - Dudley syndrom - uttalt muskelsvakhet - kan behandles med fysioterapi. Slik behandling rettet mot å styrke muskler kan være smertefullt for pasienten.
Spesielt små barn nekter ofte terapi på grunn av smertene. Til tross for intensiv trening, fører ikke fysioterapi ikke alltid til ønsket suksess. Det er likt med taleterapistøtten fra Allan - Herndon - Dudley-pasienten. Selv om reduksjonen i språkevnen kan forbedres ved intensiv trening, fører ikke alltid behandlingen til suksess på grunn av den mest høye grad av mental svekkelse av de berørte.
Frustrasjon fra pasientens side og en stor belastning for hele familien er en av de alvorligste komplikasjonene i behandlingen av Allan - Herndon - Dudley syndrom. Muskelkramper og bevegelser i ekstremitetene som ikke kan påvirkes, kan behandles med administrering av muskelavslappende midler. Komplikasjoner kan sees i noen ganger alvorlige bivirkninger av medisinene.
Utmattethet, en generell følelse av utmattelse og ubehag skal nevnes i tillegg til stress på mage-tarmkanalen. Langvarig bruk av relanxants skader også leveren og nyrene. Hvis Allan - Herndon - Dudley syndrom ikke blir behandlet, vil de berørte ikke være i stand til å gjøre noen betydelig fremgang med tanke på deres mentale eller motoriske ferdigheter.
Når bør du gå til legen?
I mange tilfeller er ikke direkte behandling av Allan-Herndon-Dudley syndrom mulig. Av denne grunn er behandlingen hovedsakelig symptomatisk og er rettet mot individuelle klager og forsinkelser. Som regel skal foreldre deretter oppsøke lege hvis barnet har muskelsvakhet.
Dette kan merkes gjennom utmattelse eller vedvarende tretthet. Medisinsk råd er også nødvendig hvis Allan-Herndon-Dudley syndrom bremser mental og motorisk utvikling.
Hvis behandlingen ikke finner sted i barndommen, kan det føre til betydelig ubehag og restriksjoner i voksen alder. En lege bør konsulteres, spesielt hvis pasienten ikke lenger kan snakke. Behandling er også nødvendig for muskelkramper. Hvis det er en akutt krise, kan du gå direkte til sykehuset eller ringe ambulanse.
I de fleste tilfeller vil Allan-Herndon-Dudley syndrom bli behandlet av en allmennlege eller av en barnelege. Imidlertid må de enkelte klagene undersøkes og behandles av den respektive spesialisten eller terapeuten.
Leger og terapeuter i ditt område
Behandling og terapi
AHDS er en årsakelig ubehandelig sykdom. Siden det ikke er noen behandlinger tilgjengelig for å rette opp den primære årsaken, har sykdommen så langt ikke vært helbredelig. I mellomtiden antyder fremskritt innen genterapi at genterapimetoder snart vil bli godkjent for daglig klinisk praksis.
I hvilken grad pasienter med syndromet vil ha fordel av godkjenning er ennå ikke avklart. For øyeblikket er det heller ikke noe etablert eller standardisert behandlingsalternativ for pasienter med AHDS innen symptomatisk terapi. For noen år siden vurderte forskere administrasjonen av TRIAC som et mulig symptomatisk terapimulighet.
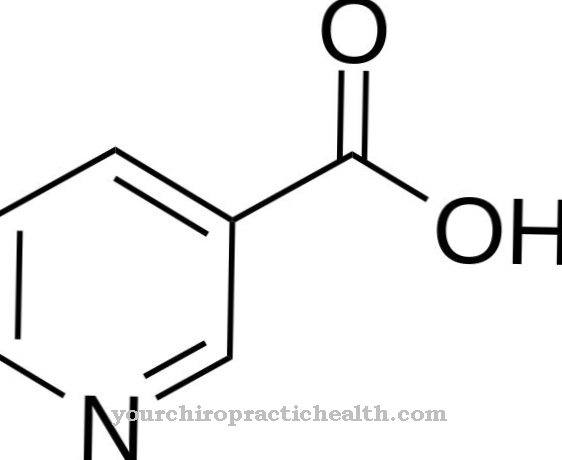
TRIAC er et ikke-klassisk skjoldbruskhormon, triiodothyroacetate. Administrering av hormonet ble utført i en klinisk studie på berørte barn, men kunne ikke oppnå noen synlige resultater. Resultatene fra studien er ikke nødvendigvis meningsfylt fordi administrasjonen av hormonet begynte relativt sent.
Av denne grunn ble TRIAC fortsatt i 2014 ansett for å være den best mulige terapien. I ett tilfelle i 2014 ble det dokumentert en betydelig forbedring i motorisk og mental utvikling under behandling med TRIAC. Terapien ble startet på den berørte personen i tidlig spedbarnsalder.
Resultatene fra hittil i studiene indikerer således at tidspunktet hvor behandlingen startes har en effekt på terapiresultatene som ikke bør undervurderes for pasienter med AHDS. Ledsagende støttende terapier som ergoterapi og fysioterapi eller tidlig intervensjon kan teoretisk sett brukes til å forbedre livskvaliteten og ferdighetene til pasientene. Imidlertid er det knapt noen bevis for effektiviteten av en slik tilnærming i forbindelse med AHDS-pasienter.
Outlook og prognose
Allan-Herndon-Dudley syndrom forårsaker en rekke forskjellige symptomer hos de fleste pasienter. Først og fremst lider de berørte av alvorlig muskelsvakhet. Dette betyr at normale aktiviteter eller idrett ikke lenger kan utføres enkelt for den det gjelder. Det er også store forsinkelser i intellektuell og mobil utvikling. Pasientens konsentrasjon er tydelig begrenset og redusert.
Det er fortsatt sterke kramper i musklene og dermed ofte ufrivillige bevegelser eller rykninger. Når Allan - Herndon - Dudley syndrom utvikler seg, kan de berørte ikke lenger snakke. Pasientens hverdag er betydelig begrenset av syndromet, og livskvaliteten reduseres. I noen tilfeller er pasientene da avhengige av hjelp fra andre mennesker i hverdagen.
Det er vanligvis ikke mulig å behandle Allan - Herndon - Dudley syndrom årsakssammenheng. Av denne grunn er behandlingen bare symptomatisk. De berørte er avhengige av forskjellige behandlingsformer, som imidlertid ikke alltid fører til et positivt forløp av sykdommen. I noen tilfeller begrenser Allan - Herndon - Dudley syndrom forventet levealder for de berørte.
Du finner medisinene dine her
➔ Medisiner for muskelsvakhetforebygging
AHDS kan bare forhindres gjennom genetisk rådgivning. Mutasjonsbærere kan for eksempel bestemme seg for å få egne barn.
ettervern
Behovet for oppfølging av genetisk forårsaket Allan - Herndon - Dudley syndrom berører bare mannlige spedbarn. Problemet er at det ikke er noen passende form for terapi for denne arvelige tilstanden. De alvorlige konsekvensene av defekter i skjoldbruskkjertelhormonsendere kan vanskelig forbedres. Forsøk på å gi de berørte barna lettelse ved å gi dem spesielle skjoldbruskhormoner har mislyktes.
Problemet er at grunnlaget for sykdommen vanligvis allerede er etablert i mors kropp. De skader det ufødte barnet permanent. Fra dette synspunktet begynner behandlingen for sent, nemlig først etter fødselen. I ettervern kan bare skaden som allerede er til stede behandles. Imidlertid er det håp. I 2014 ble en sak kjent der et spedbarn med Allan-Herndon-Dudley-syndrom ble behandlet med TRIAC. Oppfølgingsomsorg var fremdeles nødvendig fordi barnet ikke kunne bli kurert. I det minste ble symptomene hans lindret.
Allan-Herndon-Dudley syndrom er delvis forårsaket av en mangelfull blod-hjerne-barriere. Dette er hva studier ved Cedars Sinai Hospital antyder. Skjoldbruskhormonene kan ikke fungere på grunn av den mangelfulle blod-hjerne-barrieren. Dette gjelder også skjoldbruskhormoner som administreres etter at barnet er født. Bioteknologi eller genetisk forskning kan være i stand til å hjelpe. Alle forsøk på behandling mislykkes foreløpig. Dette påvirker også oppfølgingsomsorgen for hardt skadede barn.
Du kan gjøre det selv
Allan-Herndon-Dudley syndrom er en alvorlig tilstand som ennå ikke er behandlet effektivt. Foreldre kan imidlertid fortsatt gjøre noen tiltak for å støtte terapi.
For det første er regelmessig kognitiv trening og fysisk aktivitet viktig. En omfattende terapi, som avhengig av alvorlighetsgraden av syndromet, kan bestå av tale- og leseopplæring, men også av generelle hjerneøvelser, også tilbyr øvelser fra fysioterapi. Treningen må være individuelt tilpasset symptomene. Foreldrene til berørte barn bør derfor sørge for at tiltakene blir valgt optimalt og at barnet ikke blir overveldet.
Ved alvorlige psykiske lidelser kan barnet trenge permanent støtte i hverdagen. En poliklinisk omsorgstjeneste kan være en viktig lettelse for foreldrene. Døgnbehandling er like viktig, som kan støttes hjemme ved regelmessig overvåking av symptomer.
Foreldre bør også dra nytte av psykologisk rådgivning og om nødvendig også gå til en selvhjelpsgruppe, fordi kontakt med andre berørte gjør det lettere å takle sykdommen. I tillegg får foreldre ofte viktige tips for hvordan de skal takle et sykt barn.








.jpg)