Av Andersens sykdom er en spesielt alvorlig form for glykogenlagringssykdom. Det er en arvelig tilstand som er preget av dannelse av unormalt glykogen. Prognosen for sykdommen er veldig dårlig.
Hva er Andersens sykdom?

© ag visuell - stock.adobe.com
I sammenheng med Andersens sykdom en uvanlig form for glykogen lagres. Dette glykogenet har en lignende struktur som amylopektin, hvorav en høy prosentandel finnes i vegetabilsk stivelse. Vanligvis er glykogen sterkt forgrenet. I Andersens sykdom er det imidlertid bare et svakt forgrenet polysakkarid.
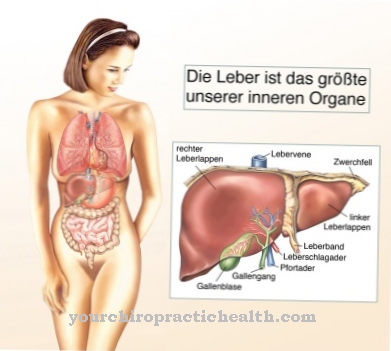
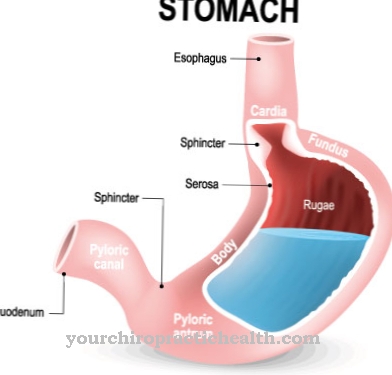
Sykdommen er preget av rask utvidelse av leveren, som raskt fører til levercirrhose. Det unormale polysakkaridet kan ikke lenger brytes ned og fortsetter å samle seg. Mangelen eller til og med mangelen på enzymet amylo-1,4-1,6-transglukosidase er ansvarlig for den mangelfulle glykogendannelsen. Det gir forgreningen i dette polysakkaridmolekylet.
Sykdommen er svært sjelden, men forekommer fortsatt i forskjellige former eller former. I den ekstremt alvorlige formen er barnet ofte dødfødt. Lettere former som begynner på senere alder er også blitt beskrevet. I alle fall er det imidlertid en mutasjon i genet (GBE1), som er lokalisert på kromosom 3.
fører til
Årsaken til Andersens sykdom er en genetisk defekt i genet GBE1 på kromosom 3, som kan arves som en autosomal resessiv egenskap. Dette genet er ansvarlig for syntesen av enzymet amylo-1,4-1,6-transglukosidase. Hvis dette enzymet mangler, eller hvis det bare har begrenset funksjonalitet, kan normalt glykogen ikke lenger syntetiseres. Enzymet er ansvarlig for forgreningen av polysakkarosemolekylet.
Hvis denne forgreningen ikke finner sted, eller hvis den bare utføres ufullstendig, opprettes et glykogen som ikke lenger kan brytes ned for en rask energiforsyning. Tvert imot, det akkumuleres veldig raskt i leveren, milten og lymfeknuter. Etter hvert måltid fraktes noe av den ubrukte glukosen til leveren for å lagre den som et reservestoff, glykogen.
Dette reservematerialet kan imidlertid ikke brukes i sin nåværende form. Den konstante ansamlingen av det unormale glykogen forstørrer leveren og milten mer og mer og fører uunngåelig til ødeleggelse av begge organer.
Symptomer, plager og tegn
Andersens sykdom manifesterer seg gjennom en ekstraordinær variabilitet. Det handler om konstant lagring av et unormalt glykogen, som ikke lenger kan brytes ned. Men alvorlighetsgraden av sykdommen kan være forskjellig. Likevel er prognosen for Andersens sykdom generelt sett svært dårlig. Det mest fremtredende symptomet er en stadig forstørrende lever, hvorfra levercirrhose raskt utvikler seg.
Den alvorligste formen viser seg gjennom manglende eller reduserte barnebevegelser før fødselen. Fosteret viser tegn på leddstivhet og lungehypoplasi. I disse tilfellene blir barnet vanligvis død. I de klassiske tilfellene er barnet fortsatt normalt utviklet ved fødselen. I de første månedene av livet utvikler imidlertid hepatomegali (forstørret lever) og hypotoni (mangel på muskelspenning).
Totalt sett er barnets utvikling forsinket. Sykdommen utvikler seg raskt. Leveren utvikler skrumplever. Det er også økt portaltrykk og milten forstørres. På grunn av skrumplever i leveren utvikler det seg varier i spiserøret med tilsvarende blødning og ascites. Døden forekommer vanligvis i tidlig barndom. I sjeldnere tilfeller starter sykdommen senere og viser symptomer på muskelsvakhet og hjertesvikt. Nevrologiske symptomer forekommer også her.
Diagnose og sykdomsforløp
Diagnosen kan stilles på grunnlag av det kliniske bildet og ledsages av laboratorietester, leverbiopsier og molekylærgenetiske tester. I de histologiske undersøkelsene merkes den intracellulære akkumuleringen av farlige amylopektinlignende strukturer. Enzymet som er ansvarlig blir undersøkt i hepatocytter, fibroblaster og leukocytter. En påvist mangel på amylo-1,4-1,6-transglukosidase bekrefter diagnosen.
komplikasjoner
Som regel reduseres forventet levealder for barnet av Andersens sykdom, eller barnet blir født død. Dette kan føre til alvorlige psykologiske plager eller depresjoner, spesielt med pårørende eller foreldre. I de fleste tilfeller er de da avhengige av psykologisk behandling.
Berørte barn lider av skrumplever i leveren, noe som til slutt fører til død. Videre er leddene også stivne og bevegelser er ikke lenger mulig på grunn av denne klagen. Barnets mentale utvikling er også sterkt svekket av Andersens sykdom, slik at de som rammes alltid alltid er avhengig av hjelp fra andre mennesker. Det er ikke uvanlig at hjertesvikt eller muskelsvakhet oppstår.
Pasientene kan også dø av hjertedød. Dessverre kan ikke Andersens sykdom helbredes. Transplantasjon av en lever kan også bare lindre symptomene i kort tid, siden skaden på den nye leveren også vil oppstå. Dette fører til slutt til barnets død. Inntil da kan imidlertid klager og symptomer begrenses ved hjelp av medisinske tiltak.
Når bør du gå til legen?
Andersens sykdom er en genetisk sykdom som i alvorlige tilfeller kan føre til død av fosteret i livmoren. Derfor bør gravide søke medisinsk behandling så snart uregelmessigheter eller unormalt blir lagt merke til under graviditet. Hvis den vordende moren har en vag følelse av at noe kan være galt med det ufødte barnet, bør hun oppsøke lege. Hvis den nyfødte overlever de første dagene og ukene etter fødselen, er det nødvendig med lege så snart særegenheter blir tydelig i det videre utviklingsforløpet. Hvis du har muskelsvakhet eller bevegelsesforstyrrelser, bør du oppsøke lege.
Forstyrrelser i vekst er tegn på en eksisterende sykdom og må avklares. Hjerteavvik, deformasjoner av kroppen og avvik i barns atferd må undersøkes og behandles. I mange tilfeller fører sykdommen til en utvidelse av organene. Spesielt leveren eller milten påvirkes i disse tilfellene.
Derfor er det nødvendig med lege så snart en uvanlig form på overkroppen oppstår i direkte sammenligning med babyer eller barn på samme alder. Misfarging av huden eller andre uregelmessigheter i hudens utseende er ytterligere tegn på helseforringelse. Et gulaktig ansikt eller øyne bør vurderes av en lege.
Terapi og behandling
Siden sykdommen er genetisk, kan ingen kausal behandling gis. Terapi er bare symptomatisk. Som en del av behandlingen fokuserer legene hovedsakelig på komplikasjoner som oppstår. Dette senker trykket i portvenes krets. Det er også en erstatning av albumin og koagulasjonsfaktorer.
Hvis leversvikt oppstår, kan en levertransplantasjon forlenge levetiden. Imidlertid kan sykdommen ikke kureres selv med en levertransplantasjon. Den genetiske defekten er til stede og vil også føre til avsetninger av det unormale glykogenet i den nye leveren. Lagring av det defekte polysakkaridet fortsetter i de andre organene i det såkalte retikulohistiocytiske systemet i milten og lymfeknuter, slik at alvorlige komplikasjoner fremdeles kan oppstå selv etter en vellykket levertransplantasjon.
Det retikulohistiocytiske systemet er en del av immunforsvaret og inkluderer cellene i det retikulære bindevevet. Disse cellene lagrer partikler og stoffer for å bryte dem ned og deretter føre dem ut av kroppen. Imidlertid er ikke nedbrytningen av de defekte polysakkarosemolekylene heller ikke lenger mulig her.
Outlook og prognose
Andersens sykdom har en relativt dårlig prognose. Den metabolske sykdommen har foreløpig ikke vært kurerbar og forårsaker alvorlig leverskade. I noen tilfeller forekommer muskelplager og samtidig sykdommer som, hvis de ikke blir behandlet, gradvis utvikler seg. Forventet levealder er sterkt begrenset av tilstanden. Syke barn når i gjennomsnitt to til fem år. En tidlig levertransplantasjon forbedrer prognosen. Prognosen er dårlig, spesielt for de klassiske sykdommens former, spesielt hvis det ikke er noen levertransplantasjon de første månedene av livet.
Som regel er den langsiktige prognosen basert på omfanget, alvorlighetsgraden og progresjonen av sykdommen. Andersens sykdom er en av de alvorligste glykogenosene. Livskvaliteten er vanligvis sterkt redusert på grunn av leverproblemer og andre symptomer. Smerte medisiner og omfattende terapi forbedrer barnets trivsel, men de er også forbundet med risiko. Leveransespesialist ansvarlig gir prognosen.
Forventet levealder er sterkt begrenset av tilstanden. Eventuelle samtidig sykdommer som kan oppstå med uoppdagede sykdommer er også inkludert i prognosen. Andersens sykdom gir derfor en dårlig prognose totalt sett. Nye behandlingsmetoder kan gi en forbedring i fremtiden.
forebygging
En forebygging av Andersens sykdom kan bare referere til det faktum at avkommet ikke arver denne sykdommen. Siden Andersens sykdom overføres på en autosomal resessiv måte, kan flere generasjoner hoppes over i arv. Hvis det allerede har vært tilfeller av Andersens sykdom i familien og pårørende, bør menneskets genetiske tester derfor utføres.
Hvis genet finnes hos begge foreldrene, anbefales genetisk rådgivning. I dette tilfellet har avkommet 25 prosent sjanse for å utvikle Andersens sykdom.
ettervern
Siden Andersens sykdom ikke kan kureres, er behandlingen av symptomene og inneslutningen av mulige komplikasjoner hovedfokuset i hele behandlingsperioden. Oppfølging er nødvendig etter inngrep som utføres som en del av terapien. Hvis det oppstår en levertransplantasjon, er profesjonell oppfølging veldig viktig for det.
Etter inngrepet sikrer dette at den nye leveren ikke blir avvist av kroppen. Spesielle medikamenter demper kroppens immunrespons. Som et resultat svekkes imidlertid kroppens motstand mot patogener, noe som må tas i betraktning i videre terapi. I løpet av denne tiden må pasienten gjennomføre regelmessige blodprøver. Det sørges for at det ikke er avvisningsreaksjoner eller andre alvorlige komplikasjoner som nyresvikt, som kan oppstå som bivirkninger.
Mens kjernesymptomene på Andersens sykdom kan forbedres direkte etter en levertransplantasjon, fortsetter avsetningen av det mangelfulle glykogenet, slik at komplikasjoner og progressive symptomer må forventes selv etter en transplantasjon. Leveransespesialist ansvarlig kan gi mer detaljert informasjon om prognosen og det videre behandlingsforløpet.
Du kan gjøre det selv
Selvhjelpstiltakene en pasient med Andersens sykdom kan ta er begrenset til ikke-eksisterende. Siden sykdommen har genetiske årsaker og ikke kan kontrolleres til tross for symptomatisk behandling, blir mulighetene til den som blir rammet raskt oppbrukt. Han anbefales best å ta ethvert kostholds- og livsstilsråd gitt av sin behandlende lege på alvor og å implementere det.
Videre, etter en levertransplantasjon, bør personen som rammes vurdere mild oppførsel. Alkohol, fet mat og anstrengelse bør unngås. Dette gjør det lettere for kroppen å virkelig godta det nye orgelet. Imidlertid kan en vellykket transplantasjon, inkludert vellykket oppfølging, ikke stoppe glykogenose av type 4 selv.
Siden dette er en autosomal recessiv arvelig sykdom (den kan hoppe over flere generasjoner), er det fornuftig å få utarbeidet en genetisk profil når du planlegger en familie. Mens de som er rammet av Andersens sykdom allerede vet om genet sitt, er en analyse i denne forbindelse spesielt verdt for familiemedlemmer. På denne måten kan overføringen av det utløsende genet forhindres gjennom passende familieplanlegging. I det minste kan det imidlertid oppnås en visshet om risikoen for sykdom hos ens eget avkom.

.jpg)







.jpg)
.jpg)
.jpg)
.jpg)