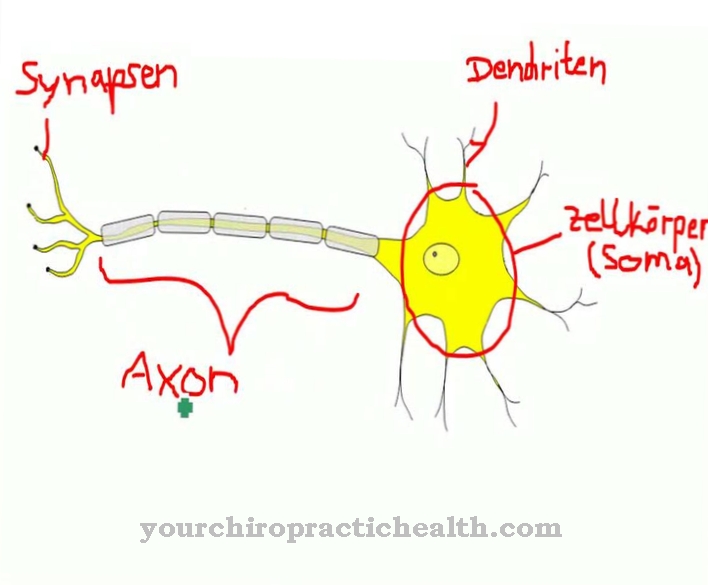
På α-N-acetylgalactosaminidase mangel det er en lysosomal lagringssykdom som forekommer svært sjelden. Det er delt inn i en ung og en voksen form.
Hva er a-N-Acetylgalactosaminidase-mangel?

A-N-acetylgalactosaminidase-mangelen eller Alpha-N-acetylgalactosaminidase mangel kalles også i medisin Schindlers sykdom, Schindlers sykdom eller Kanzakis sykdom utpekt. Det som menes er en ekstremt sjelden lysosomal lagringssykdom, der arven er autosomal recessiv. Leger skiller mellom en ung form, som forekommer hos barn og unge, og en voksen form, som voksne lider av.
Den unge formen for alfa-N-acetylgalaktosaminidase-mangel kalles Schindlers sykdom, mens den voksne formen kalles Kanzakis sykdom. Men det er også underinndelinger i Schindlers sykdom type I for ungdommelig form og Schindlers sykdom type II for voksenform. En blanding av begge former, kjent som Schindlers sykdom type III, er også mulig.
Alle tre former for sykdommen er oligosakkaridoser (glykoproteinoser), som inkluderer fucosidose og lagring av sialinsyre. Navnet Schindlers sykdom eller Schindlers sykdom for alfa-N-acetylgalaktosaminidasemangel går tilbake til den tyske genetikeren Detlev Schindler.
Han var den første som beskrev sykdommen i 1988. Et år senere beskrev den japanske legen T. Kanzaki voksenformen. I 1991 oppdaget han at en alfa-N-acetylgalaktosaminidase-mangel var årsaken til sykdommen.
fører til
Α-N-acetylgalactosaminidase-mangelen er forårsaket av en redusert aktivitet av enzymet alfa-N-acetylgalactosaminidase. Missense eller tullmutasjoner på NAGA-genet, som koder for alfa-N-acetylgalaktosaminidase, er ansvarlige for enzymdefekten.
NAGA-genet er lokalisert på kromosom 22-genlokus q11. Enzymet alpha-N-acetylgalactosaminidase har som oppgave å katalysere spaltningen av N-acetylgalactosamine fra forskjellige glykolipider og glykoproteiner. Den genetiske defekten forårsaker en redusert aktivitet av enzymet, slik at stoffer som ikke metaboliseres samler seg i kroppens celler.
I de fleste tilfeller er dette proteoglykaner, glykosfingolipider og N- eller O-koblede glykoproteiner. Opphopningen av disse forårsaker skade på cellen og de berørte organene. Alpha-N-acetylgalactosaminidase-mangel er så sjelden at det ikke er presis informasjon om frekvensen.
Så langt er det bare tolv pasienter fra åtte familier som er kjent for å ha utviklet Schindlers sykdom i verden. Nevrologiske klager forekommer ikke hos alle pasienter. Noen leger mistenker påvirkning av flere faktorer for forekomst av nevrologiske symptomer. Det er imidlertid fremdeles ingen bevis som støtter denne antagelsen.
Du finner medisinene dine her
➔ Medisiner mot smerterSymptomer, plager og tegn
Symptomene på alpha-N-acetylgalactosaminidase-mangel avhenger av den spesielle sykdomsformen. I sammenheng med Schindlers sykdom type I, oppstår progressiv muskelhypotoni det første leveåret. I tillegg lider de berørte babyene av uttalt psykomotorisk regresjon.
Forstyrrelser i bevegelsessekvensen, spastisk tetraplegi og myoklonale cerebrale anfall ble også registrert som symptomer. Det er også en risiko for at barnet blir blind. Hvis det er en voksen form for alpha-N-acetylgalactosaminidase-mangel, dvs. Schindlers sykdom type 2, oppstår symptomer som ligner på Fabrys sykdom.
Pasienter har angiokeratomer, som er godartede hudforandringer. Mange pasienter er svakt utviklingshemmede. En klinisk mellomform er Schindlers sykdom type III De rammede lider av nevrologiske funksjonssvikt, hjerneanfall og psykiske funksjonshemninger.
Mindre alvorlige former er imidlertid også mulige, som er assosiert med milde psykiatriske og nevrologiske plager, forsinket språkutvikling eller symptomer som ligner autisme.
Diagnose og kurs
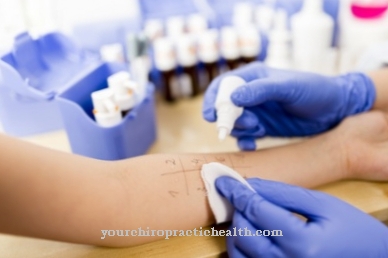
Diagnosen en alpha-N-acetylgalactosaminidase-mangel er mulig gjennom påvisning av redusert NAGA-aktivitet. For dette formål blir enzymprøver utført i blodplasma, leukocytter eller dyrkede fibroblaster eller lymfoblaster. Den kromatografiske deteksjonen av oligosakkarider i urinen muliggjør også diagnosen enzymmangel.
En DNA-analyse kan også utføres, men dette er ikke nødvendig i de fleste tilfeller. Under graviditet kan også prenatal diagnose av enzymmangel utføres ved bruk av en mutasjonsanalyse av NAGA-genet etter en kororisk villusprøvetaking eller fostervannsprøve. Imidlertid kan ikke den respektive sykdomsformen bestemmes.
Den differensielle diagnosen Fabry syndrom, panthenat kinase-assosiert nevrodegenerasjon og infantil neuroaxonal dystrofi, som også har enzymdefekter, er også viktig. Forløpet av alpha-N-acetylgalactosaminidase-mangelen avhenger av den respektive sykdomsformen.
Schindlers sykdomstype I regnes som ugunstig, mens prognosen for de to andre typene er gunstigere. Antall syke mennesker er imidlertid så lite at det ikke kan komme noen meningsfulle uttalelser om forventet levealder.
komplikasjoner
Symptomene og komplikasjonene ved A-N-acetylgalactosaminidase-mangel avhenger sterkt av dens form.I de fleste tilfeller forekommer imidlertid bevegelsesforstyrrelser, slik at de berørte er relativt begrenset i hverdagen. Kramper forekommer også, som er assosiert med sterke smerter.
Spesielt babyer lider av alvorlige utviklingsforstyrrelser, noe som kan føre til spastisitet hos barnet. I noen tilfeller fører A-N-acetylgalactosaminidase-mangelen til fullstendig blindhet hos pasienten. Regresjonen av intelligens fører til utviklingshemning og dermed til funksjonshemninger.
Disse begrenser vanligvis pasientens hverdag betydelig og har en negativ innvirkning på livskvaliteten. Ofte er de berørte avhengig av hjelp fra andre mennesker. Retardasjonen kan også føre til et ordfunnsforstyrrelse eller en språkforstyrrelse. Det er ikke mulig å behandle A-N-acetylgalactosaminidase-mangelen kausalt.
Av denne grunn må den berørte personen ta antibiotika. Visse komplikasjoner må også unngås direkte. For eksempel lungebetennelse er en av disse. Videre kan pasienten også være avhengig av kunstig ernæring.
Når bør du gå til legen?
A-N-acetylgalactosaminidase-mangel må definitivt behandles av lege. Denne mangelen forsvinner ikke av seg selv, og det er ingen spontan helbredelse av denne sykdommen. Uansett må en lege konsulteres hvis foreldrene merker en nedgang i motoriske og mentale evner hos barnet eller babyen. Klager med enkle bevegelser eller prosesser kan også indikere A-N-acetylgalaktosaminidasemangel og bør undersøkes av lege.
Videre er forskjellig spastisitet også et tegn på en sykdom. Det er ikke uvanlig at pasienter lider av intellektuell funksjonshemning og andre funksjonsfeil. Hvis disse klagene er til stede, er behandling definitivt nødvendig. Hvis det ikke er noen behandling, kan dette føre til betydelige klager og komplikasjoner i voksen alder og dermed redusere livskvaliteten til den som blir rammet betraktelig.
Alvorlig forsinket taleutvikling kan også være et tegn på A-N-acetylgalaktosaminidase-mangel og må derfor undersøkes. Som regel kan en allmennlege konsulteres for å bestemme A-N-acetylgalaktosaminidase-mangelen. Den videre behandlingen av de enkelte klagene blir deretter utført av en spesialist.
Leger og terapeuter i ditt område
Behandling og terapi
En kausal terapi for alfa-N-acetylgalaktosaminidase-mangel er ikke mulig. Derfor er den medisinske tilnærmingen begrenset til å behandle symptomene. Dette inkluderer et fornuftig kosthold og væskeinntak, forebygging av smittsomme sykdommer, som også kan gjøres ved å administrere antibiotika, samt å dempe hjerneanfall ved å administrere antiepileptika.
Videre behandlingstrinn inkluderer fysioterapitiltak for å forhindre lungebetennelse og kontrakturer, og for å redusere smerter eller spastisitet ved hjelp av medisiner. Videre kan aspirasjonsprofylakse skje gjennom kunstig fôring.
Nyere studier har vist at a-N-acetylgalactosaminidase-mangelen er en proteinfoldingsforstyrrelse, slik at enzymerstatningsterapi blir diskutert som et fornuftig behandlingstiltak. Genterapi er en annen tenkelig terapeutisk tilnærming for behandling av lysosomale lagringsforstyrrelser. Fordi Schindlers sykdom er arvelig på en autosomal resessiv måte, anbefales genetisk rådgivning av de berørte familiene.
Outlook og prognose
A-N-acetylgalactosaminidase-mangelen kan føre til forskjellige symptomer. I de fleste tilfeller fører imidlertid mangelen til forsinket psykomotorisk utvikling. I voksen alder er pasienten ofte avhengig av hjelp fra andre mennesker og kan ikke takle hverdagen på egen hånd. Bevegelsesforstyrrelser forekommer også. Dette kan bli lagt merke til ved en halte eller av forstyrrelser i koordineringen.
I verste fall kan pasienten bli blind eller lide av alvorlige synsforstyrrelser på grunn av A-N-acetylgalactosaminidase-mangel. Det er ikke uvanlig at det oppstår intellektuelle funksjonshemninger som reduserer livskvaliteten til den aktuelle personen i stor grad. Taleforstyrrelser og ordfunnsforstyrrelser forekommer også, noe som kan påvirke livet. Spesielt barn rammes av mobbing og erting på grunn av symptomene på A-N-acetylgalactosaminidase-mangel.
En kausal behandling av A-N-acetylgalactosaminidase-mangelen er ikke mulig, slik at behandlingen først og fremst er rettet mot å redusere symptomene. Ulike terapeutiske tiltak kan brukes for å redusere utviklingsforstyrrelser. I mange tilfeller er imidlertid pasienten avhengig av hjelp utenfra i hverdagen. Forventet levealder påvirkes ikke av mangelen.
Du finner medisinene dine her
➔ Medisiner mot smerterforebygging
Det er ingen kjente forebyggende tiltak mot alfa-N-acetylgalaktosaminidasemangel. Schindlers sykdom er en av de ekstremt sjeldne sykdommene.
ettervern
Siden A-N-acetylgalactosaminidase-mangel er en medfødt sykdom som ikke kan behandles årsakssammenheng, men bare symptomatisk, er fullstendig behandling ikke mulig. Den berørte personen er avhengig av livslang behandling, og derfor er alternativene for ettervern veldig begrenset.
På grunn av mangel på A-N-acetylgalactosaminidase er pasienten ofte avhengig av inntak av medisiner og antibiotika. Eventuelle interaksjoner med andre medisiner bør tas i betraktning, selv om antibiotika ikke bør tas sammen med alkohol. Hvis du har et angrep, bør du gå til sykehuset eller ringe en legevakt direkte.
Videre bør lungene skånes for å unngå betennelse. Pasienter med A-N-acetylgalactosaminidase-mangel bør derfor ikke røyke under noen omstendigheter. Et sunt kosthold og en generelt sunn livsstil har en veldig positiv effekt på sykdomsforløpet.
Hvis pasienten ønsker å få barn, er genetisk rådgivning nyttig for å forhindre at sykdommen blir overført til barna. Siden pasientene ofte lider av begrenset mobilitet, er fysioterapi nyttig for å lindre dette. Mange øvelser kan også utføres i ditt eget hjem.
Du kan gjøre det selv
En eksisterende A-N-acetylgalactosaminidase-mangel er en uhelbredelig sykdom. Medisinsk behandling er derfor viktig. Selvbehandlingstiltak kan bare baseres på symptomene for å gjøre hverdagen enklere. Det er viktig at foreldrene følger barnet nøye.
Foreldre kan best hjelpe barnet sitt gjennom forståelse og tålmodighet, kjærlighet og omsorg. Videre er ergoterapi- og logopeterapi alltid basert på et dobbelt konsept: terapi på stedet med spesialisten og utførelse av øvelsene hjemme. Foreldre bør være aktive her konsekvent og med tålmodighet. Det må sikres et kosthold rikt på vitaminer og mineraler, regelmessig mosjon og rikelig med frisk luft. Dette styrker immunforsvaret og reduserer risikoen for infeksjon. Hvis personen har en tendens til å gripe, bør de ikke være alene på lenge og de bør sjekkes for å se om de kan skade seg selv hvis et anfall oppstår.
Det meste av tiden klarer ikke pasienter å takle hverdagen selv og krever konstant omsorg. Hvis foreldre ikke har råd til dette - spesielt ikke med eldre barn og voksne - skal de ikke være redde for å søke hjelp. Dette kan ha form av en sykepleier eller plassering i et tilstrekkelig anlegg. Hvis du fortsatt ønsker å få barn, anbefales en konsultasjon med en spesialist, siden sykdommen er arvet.
.jpg)









.jpg)
.jpg)
.jpg)
.jpg)