De Lipoprotein lipase (LPL) hører til lipasene og spiller en avgjørende rolle i lipidmetabolismen. Det er ansvarlig for å dele triglyseridene i kylomikronene og lipoproteiner med veldig lav tetthet (VLDL) i fettsyrer og monoacylglyserol. De frigjorte fettsyrene brukes til å generere energi eller til å bygge opp kroppsfett.
Hva er lipoprotein lipase?
Lipoprotein lipase (LPL) er et enzym som er en av lipasene. Lipaser er ansvarlige for å bryte ned triglyserider (triacylglyseroler) til fettsyrer og glyserin. Triglyserider er estere av trippelalkoholglyserin med tre fettsyrer hver. De er kjent som fett eller fettoljer.
Kostholdsfett blir absorbert med mat og blir først brutt ned av de ekstracellulære lipaser fra bukspyttkjertelen i tarmen. Noen triglyserider kommer imidlertid inn i blodomløpet gjennom serumet når de blir absorbert i tynntarmen, hvor de er bundet til lipoproteiner, noe som garanterer deres transportevne i blodet. Lipoprotein lipase er enzymet som bryter ned triglyseridene bundet til lipoproteinene til fettsyrer og monoacylglycerol. Den består av 448 aminosyrer og er avhengig av koenzym apolipoprotein C2 for sin funksjon.
Lipoprotein lipase er et vannløselig enzym som er bundet til endotelcellene i blodkarene via visse glykoproteiner (proteoglycans). Det produseres i leveren. Enzymet katalyserer hydrolysen av triglyseridene for å danne to fettsyremolekyler og ett monoacylglyserolmolekyl. Apolipoproteinene er bærermolekylene til triglyserinene og gjør det mulig å transportere dem i et vandig miljø. Apolipoprotein C2 fungerer også som reseptor for lipoprotein lipase og aktiverer dermed hydrolysen av triglyseridene.
Funksjon, effekt og oppgaver
Funksjonen til lipoprotein lipase er å fullstendig katalysere nedbrytningen i blodet til fett som er absorbert av tarmcellene. For det første blir diettfettene brutt ned til fettsyrer og glyserin av pankreas lipaser i tynntarmen. Ytterligere triglyserider kommer inn i blodet gjennom absorpsjon via tynntarmen og bindes der til lipoproteiner for å danne et lipidproteinkompleks.
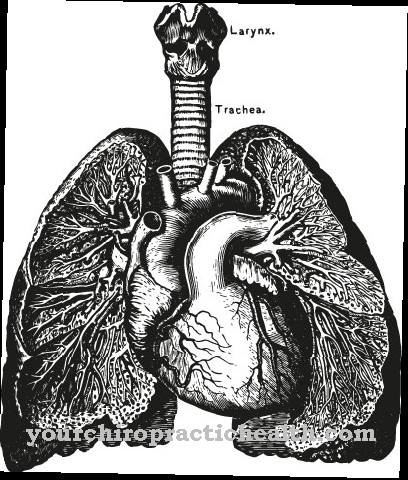
Dette skaper kylomikroner. De representerer lipoproteinpartikler med en diameter på 0,5 til 1 mikrometer. Deres tetthet er mindre enn 1000 g / ml. Lipidkjernen inneholder hovedsakelig triglyserider med en liten mengde kolesterolestere. Kolesterolholdige skall av kylomikronene inneholder fosfolipider som et strukturelt element. Apolipoproteinene som triglyseridene er bundet til, lagres nå også i dette skallet. Kylomikronene inneholder 90 prosent triglyserider. De kommer inn i blodomløpet fra tynntarmen via lymfesystemet. Triglyseridene brytes ned i fettsyrer og glyserin ved hjelp av LPL, spesielt i kapillærene i muskel og fettvev.
Fettsyrene brukes enten i muskelvev for å generere energi eller i fettvev for å bygge opp endogene triglyserider som lagringsfett. Etter omtrent ti timers avholdenhet med mat, kan ikke flereylomikroner påvises i blodet fordi triglyseridene da blir helt nedbrutt. Andre komponenter i blodet er det såkalte VLDL (lipoprotein med svært lav tetthet). Disse strukturelle enhetene frigjøres fra leveren og inneholder triglyserider, fosfolipider og kolesterol. VLDL transporterer disse komponentene gjennom blodstrømmen fra leveren til de enkelte organer.
På denne måten blir triglyseridene brutt ned av lipoprotein lipase og de frigjorte fettsyrene blir absorbert av kroppens celler. Nedgangen i triglyserider omdanner VLDL til LDL (Lipoprotein med lav tetthet). LDL inneholder hovedsakelig fosfolipider, kolesterolestere og lipoproteiner
Utdanning, forekomst, egenskaper og optimale verdier
Lipoprotein lipase syntetiseres i leveren. I tillegg til lipreasene i bukspyttkjertelen, representerer den en annen ekstracellulær lipase.LPL er plassert på utsiden av membranene til endotelceller i en lang rekke organer, inkludert fettceller. Der er den koblet til cellemembranene via såkalte proteoglykaner.
Imidlertid er det av spesiell betydning for endotelcellene i blodkarene, siden det her direkte kan kontrollere hydrolysen av triglyseridene i kylomikronene og VLDL-ene. Heparin blir injisert for å måle lipoproteaseaktivitet. Heparin fjerner bindingen av lipoprotein-lipaser fra proteoglykanene, slik at det etter en heparininjeksjon er en økt konsentrasjon av frie lipoprotein-lipaser, som kan bestemmes av deres aktivitet. Denne undersøkelsen kan blant annet bestemme en mangel på lipoprotein lipase.
Sykdommer og lidelser
Mangelen på lipoprotein lipase fører ofte til alvorlige helseproblemer. Hvis det er for lite lipoprotein lipase eller hvis aktiviteten er utilstrekkelig på grunn av en genetisk defekt, kan triglyseridene i kylomikronene og VLDL-ene bare brytes ned dårlig eller ikke i det hele tatt.
Lipoprotein lipase mangel kan være primært genetisk, så vel som sekundær til cellegift, for eksempel. Primær LPL-mangel er sjelden og er forårsaket av en autosomal recessiv genetisk defekt. En såkalt chylomicronemia utvikler seg, som er preget av et melkeaktig, kremet serum og blir referert til som type I hyperlipidemia. Triglyseridene i kylomikronene brytes ikke lenger ned. Som et resultat forekommer alvorlige pankreatider med melkeintoleranse og magesmerter igjen og igjen.
I tillegg utvikler det seg konstant sprengning av xantomer og hepatomegali. De eneste behandlingsalternativene er et lite fett kosthold og ingen alkohol. Denne sykdommen er ofte forårsaket av mutasjoner i LPL-genet på kromosom 8 eller i APOC2-genet. Den sekundære formen for hyperlipidemi av type I forekommer vanligvis med cellegift og er bare av midlertidig karakter.







.jpg)