De Mukopolysakkaridose er en samlebetegnelse for lysosomale lagringssykdommer som er basert på lagring av glykosaminoglykaner. Alle sykdommer utvikler lignende symptomer og former. Alvorlighetsgraden av syndromene varierer veldig.
Hva er mukopolysakkaridose?

© Sebastian Kaulitzki - lager.adobe.com
EN Mukopolysakkaridose det er ikke noe som heter en enkelt sykdom. Begrepet mucopolysaccharidosis er en samlebetegnelse for et stort antall lagringssykdommer.Disse er basert på lagringsforstyrrelser av glykosaminoglykaner (GAG) i lysosomene til celler. Lagringen skjer gradvis fordi terminering av tilkoblinger ikke fungerer.
Alle mukopolysakkaridoser er genetiske. Hver sykdom mangler et spesifikt enzym som katalyserer nedbrytningen av den tilsvarende GAG. Alle mukopolysakkaridoser er svært sjeldne sykdommer og viser ofte lignende forløp. Hvis de ikke blir behandlet, ødelegger de stadig økende forekomstene cellene. Organene blir ødelagt i prosessen. Sykdommen kan begynne både i spedbarnsalderen og i voksen alder.
Mukopolysakkaridose kan være forårsaket av fire forskjellige grupper av glykosaminoglykaner:
- Heparansulfat
- Keratansulfat
- Kondroitinsulfat
- Dermatan sulfat.
Alle glykosaminoglykaner består av en polysakkaridkjede festet til et protein. Karbohydratkomponenten utgjør 95 prosent og proteinkomponenten fem prosent av molekylmassen. Avhengig av hvilken glykosaminoglykan og hvilket enzym som er påvirket, kan mukopolysakkaridoser deles inn i seks forskjellige hovedformer: Disse inkluderer Hurler / Scheies sykdom (MPS I), Hunter's sykdom (MPS II), Sanfilippos sykdom (MPS III), Morquios sykdom ( MPS IV), Maroteaux-Lamys sykdom (MPS VI), og Slys sykdom (MPS VII). Alle typer har alvorlige og milde former.
fører til
Årsaken til alle mukopolysakkaridoser er en økende lagring av glykosaminoglykaner (GAGs) i lysosomene i cellene. Nedbrytningen av de tilsvarende biopolymerene forstyrres. For hver individuelle forstyrrelse mangler enten et visst enzym, eller dette enzymet fungerer feil. Det kan være flere mutasjoner for hvert enzym. Arven etter den korresponderende mutasjonen kan være autosomal recessiv, autosomal dominant eller x-bundet recessiv.
Siden en enzymatisk prosess vanligvis involverer flere reaksjonstrinn, kan flere enzymer teoretisk muteres for den samme glykosaminoglykan. Symptomene på lidelsen vil være de samme eller lignende.
- På MPS I, Hurlers eller Scheies sykdom, er enzymet alfa-l-iduronidase mangelfull.
- MPS II representerer jegersyndromet med en mangelfull iduronat-2-sulfatase.
- Sanfilippo-syndromet (MPS III) kan deles inn i flere undertyper. Flere enzymer kan påvirkes i denne tilstanden.
- Morquios sykdom (MPS IV) er forårsaket av en mangelfull ß-galaktosidase.
- I Maroteaux-Lamy syndrom (MPS VI) det er N-acetyl-galaktosamin-4-sulfat-sulfatase.
- Slys sykdom (MPS VII) er forårsaket av mangelfull β-glukuronidase. Når de tilsvarende glykosaminoglykanene lagres i lysosomene, blir disse større og større.
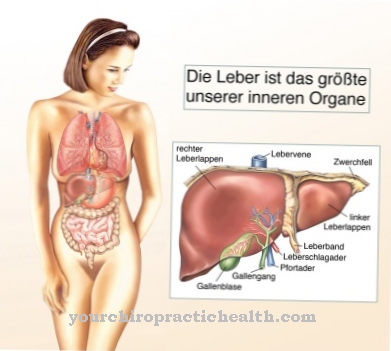
Cellene forstørres også fordi de trenger mer og mer plass for ikke-degraderte GAG-er. Dette merkes også ved utvidelsen av mange organer. Et typisk symptom er den konstante forstørrelsen av leveren og milten. Hvis de ikke blir behandlet, fører lagringssykdommene til død gjennom gradvis ødeleggelse av organene.
Symptomer, plager og tegn
Symptomene er like for alle sykdommer. Det er alvorlige og milde former. Imidlertid betyr et mildt forløp bare at sykdommen utvikler seg saktere. Det endelige kurset er alltid det samme. Det er progressiv deformasjon av skjelettsystemet, leddkontraksjoner, grov ansiktsdrag og utvidelse av leveren og milten.
De mentale og motoriske ferdighetene avtar på kort eller lang sikt. I de alvorlige formene av lidelsene er de kliniske bildene veldig like. Umbilical og inguinal hernias, hjerteproblemer og luftveisinfeksjoner oppstår på et tidlig stadium. Over tid utvikler innsnevringen av luftveiene og utvidelsen av mandlene og mandlene til enorme søvnapnéproblemer.
Diagnose og sykdomsforløp
Mukopolysakkaridoser kan diagnostiseres ved å undersøke urinen for utskilt glykosaminoglykaner. Ved mukopolysakkaridose økes alltid verdiene. Aktiviteten til det mistenkte defekte enzymet i leukocytter eller fibroblaster kan også bestemmes. Et visst mønster med utskillelse av glykosaminoglykanene fører mistanken til et tilsvarende enzym, som deretter blir undersøkt.
komplikasjoner
På grunn av mukopolysakkaridose lider de berørte av forskjellige misdannelser og skjelettplager. Deformasjoner forekommer som kan begrense pasientens hverdag betydelig. Som regel påvirkes leddene også av mukopolysakkaridose, slik at pasientens bevegelse er begrenset.
Spesielt barn blir rammet og lider av alvorlig forsinket utvikling, slik at ulike følgeskader også kan oppstå i voksen alder. Det er ikke uvanlig at mucopolysaccharidosis forårsaker problemer i hjertet eller pusten. I verste fall kan plutselig hjertedød føre til at den det gjelder. På grunn av pustevansker, lider pasienter av tretthet og tretthet.
Motstanden til de berørte synker også enormt. Det er ikke uvanlig at pustevansker om natten fører til søvnproblemer og dermed til depresjon. Livskvaliteten til pasienten reduseres betraktelig av mukopolysakkaridose. En kausal behandling av denne sykdommen er dessverre ikke mulig. De berørte er derfor avhengige av benmargsgivere for å behandle symptomene. Det er ingen spesielle komplikasjoner. Imidlertid er i de fleste tilfeller pasienter avhengige av livslang terapi.
Når bør du gå til legen?
Endringer og avvik i kroppsstrukturen indikerer en svekkelse i helsevesenet. Et legebesøk er nødvendig så snart det er permanente optiske særegenheter eller vedkommende har problemer med å bevisst optimalisere holdningen.Hevelse i leddene, endringer i ansiktstrekk eller utvidelse av brystet bør undersøkes intenst av en lege slik at en diagnose kan stilles. En lege er påkrevd hvis det er begrensninger i mulighetene for bevegelse, uregelmessigheter i hverdagen frivillig kontroll og en reduksjon i fysisk og mental ytelse. Rådfør deg med lege i tilfelle forstyrrelser i hjerterytmen, pusteproblemer eller avbrudd under nattesøvn.
Hevelse i halsen, en følelse av tetthet i halsen, forstyrrelser i svelget og endringer i vokalisering anses å være bekymringsfull. De må undersøkes av lege slik at symptomene kan lindres. Hvis vedkommende lider av flere infeksjoner, hvis evnen til å konsentrere seg og ta hensyn til nedsatt eller hvis navlestreng eller navlebrokk oppstår gjentatte ganger, bør lege informeres om observasjonene.
Plutselige hudskader, gulfarging av huden samt indre rastløshet bør undersøkes og behandles. En lege er nødvendig så snart det er smerter i kroppen, redusert livskvalitet og atferdsproblemer. Hvis det er fare for åndenød, er det nødvendig med ambulansetjeneste. For å forhindre denne akutte tilstanden, bør en lege konsulteres så tidlig som mulig.
Terapi og behandling
En kausal terapi er ikke mulig i dag ennå. Imidlertid er det noen tilnærminger til fremtidig genterapi for disse sykdommene i forskningsprosjekter. Dessverre er det foreløpig ingen konkrete resultater på dette området. Imidlertid skal en klinisk studie om genterapi for Hurlers sykdom starte i Barcelona. Med noen former for mukopolysakkaridoser har overføringer av benmarg vist seg effektive i enkelttilfeller. Dette rammer for eksempel Jegers sykdom, Hurlers sykdom eller Sanfilippos sykdom.
Gjennom denne benmargsoverføringen byttes de syke stamcellene mot sunne stamceller fra en giver. Dette gjør det mulig for organismen å gjenopprette tilstrekkelig det manglende enzymet. Enzymerstatningsterapi lønner seg også i mange tilfeller. Imidlertid må denne erstatningsterapien utføres for livet. Imidlertid er det også tilfeller der lovende terapier ikke lenger er mulig. Poenget her er imidlertid å gjennomføre symptomatiske behandlinger.
Du finner medisinene dine her
➔ Medisiner mot smerterOutlook og prognose
Den videre utviklingen hos pasienter med mukopolysakkaridose må vurderes individuelt. Dette begrepet er en samlebetegnelse på forskjellige lagringssykdommer. Disse er til stede i forskjellige grader hos hver pasient, og intensiteten er individuelt uttalt. Hvis det ikke legges inn medisinsk behandling, blir de indre organene til alle de berørte gradvis ødelagt i løpet av livet. Dette resulterer i en forkortelse av gjennomsnittlig forventet levetid.
Med en tidlig diagnose kan en personlig optimalisert terapi utarbeides. Dette er knyttet til helsekravene og eksisterende klager fra pasienten. Langtidsbehandling er grunnleggende nødvendig for å oppnå en stabil bedring i helsen. Kirurgiske inngrep kan forekomme, som hver er assosiert med forskjellige risikoer og bivirkninger. Hvis operasjonen fortsetter uten ytterligere komplikasjoner, blir symptomene vanligvis lettet etterpå.
Likevel kan uønsket utvikling og tilbakeslag oppstå i løpet av livet. I enkelttilfeller kan bare en benmargstransplantasjon forbedre den generelle livskvaliteten. På grunn av de generelle omstendighetene opplever pasienten en sterk følelsesmessig og mental belastning. Normal hverdag er ofte ikke mulig på grunn av symptomene. Psykologiske komplikasjoner kan oppstå og føre til en ytterligere forverring av situasjonen.
forebygging
Siden mukopolysakkaridoser er arvelige sykdommer, er ikke forebygging mulig. I tilfelle av en eksisterende sykdom, kan suksessen med behandlingen sikres gjennom rettidig behandling. I tillegg er konstant overvåking av lunge- og hjertefunksjonen nødvendig. Hvis tilfeller av mukopolysakkaridose allerede har oppstått i familien, kan risikoen for sykdommen vurderes gjennom genetisk rådgivning hvis familien ønsker å få barn.
ettervern
I de fleste tilfeller av mukopolysakkaridose har pasienten bare noen få alternativer for oppfølging, slik at den som rammes først og fremst bør oppsøke lege på et tidlig tidspunkt. Bare med tidlig oppdagelse og behandling av denne sykdommen kan ytterligere komplikasjoner forhindres, slik at lege bør kontaktes så snart de første tegn og symptomer vises.
I de fleste tilfeller er de berørte avhengig av kirurgiske inngrep, som kan lindre og begrense symptomene. Men siden mukopolysakkaridose er en genetisk sykdom, kan den vanligvis ikke kureres fullstendig.
Derfor bør vedkommende først oppsøke lege hvis de ønsker å få barn for å forhindre at sykdommen kommer igjen hos barna. Det er ofte veldig viktig å ha familiestøtte under behandlingen. Dette kan også forhindre depresjon og andre psykologiske opprør. Mukopolysakkaridose kan føre til redusert forventet levealder for den som blir rammet, hvorved det videre forløpet avhenger veldig av diagnosetidspunktet.
Du kan gjøre det selv
Mulighetene for selvhjelp når det gjelder mukopolysakkaridose er begrenset til å lindre symptomer og dermed forbedre livskvaliteten. Selvhjelpsgrupper har vist seg å være svært nyttige, ettersom utvekslingen med andre foreldre avslører verdifulle tips og ofte kan lindre frykt og bekymring og gi et mer positivt syn på fremtiden.
Akkompagnementet til fysioterapi, ergoterapi, logopedi og andre former for terapi, som ofte kan utdypes i hjemmemiljøet, er nå en integrert del av livet.
For å gjøre livet så enkelt som mulig for deg selv og det berørte barnet, anbefales det å gjøre bomiljøet tilgjengelig for funksjonshemmede så tidlig som mulig. Med økende alder og vekt på barnet, viser høydejusterbare pleiesenger seg å være en stor fysisk lettelse for omsorgspersonen. Varslingsapparater for epilepsi og andre tekniske hjelpemidler gir best mulig sikkerhet, selv om natten og avlaster foreldrene om natten slik at de kan sove mer avslappet.
Å føre en symptomdagbok kan hjelpe legen til å gjenkjenne nye symptomer og muligens å rette opp behandlingen av eksisterende symptomer, siden medikamentell behandling ofte ikke viser ønsket, men motsatt effekt.
Siden sykdommen er veldig krevende for pårørende, må de lage små plasser for seg selv for å lade batteriene. Dette kan omfatte kurer, forebyggende pleie eller senere ferie på hospits.
.jpg)

.jpg)
.jpg)







.jpg)
.jpg)
.jpg)
.jpg)