Av Niemann-Pick sykdom er også som Niemann-Pick sykdom kjent. Den arvelige sykdommen er en av de lysosomale lagringssykdommene.
Hva er Niemann-Pick sykdom?

© ktsdesign - stock.adobe.com
Av Niemann-Pick sykdom er en sykdom fra gruppen av sfingolipidoser. Dette er metabolske sykdommer som stort sett manifesterer seg i sentralnervesystemet. Innenfor sfingolipidosene tilhører sykdommen de lysosomale lagringssykdommene. Disse er preget av funksjonsfeil i lysosomene.
I engelsktalende land er begrepet Lysosomale lagringssykdommer (LSDer) brukes. Ved Niemann-Picks sykdom blir sfingomyelin avsatt i leveren, benmargen, milten og hjernen. Sykdommen ble oppkalt etter oppdagerne Albert Niemann og Ludwig Pick. Den ble først beskrevet i 1914. Niemann-Pick sykdom forekommer ganske sjelden.
Rundt en nyfødt hos 8000 fødsler vil utvikle en lysosomal lagringssykdom. Men det inkluderer ikke bare det Niemann-Pick sykdom, men også sykdommer som Hunter syndrom eller Sanfilippo syndrom.
fører til
Niemann-Pick sykdom er arvelig som en autosomal resessiv egenskap. Ved autosomal recessiv arv er den mangelfulle allelen på et homologt kromosom eller et autosom. Bare homozygote bærere av egenskapen blir syke. Dette betyr at det genetiske materialet i en celle må ha to identiske kopier av det defekte genet på begge kromosomene for at sykdommen skal bryte ut.
Niemann-Pick-syndromet er basert på en genetisk enzymfeil. Enzymet sfingomyelinase påvirkes. Sfingomyelinasen er ansvarlig for spaltning av sfingomyelin. Enzymdefekten fører til økt lagring av sfingomyeliner i lysosomene i milten, benmargen, hjernen og leveren. Lysosomer er celleorganeller som inneholder fordøyelsesenzymer.
De fordøyer fremmed materiale som patogener eller cellevfall. De spiller også en viktig rolle i programmert celledød (apoptose). I dyreforsøk kan det vises at ekspresjonen av den regulerende faktor for myelingen (MRF) er betydelig redusert av mutasjonen i NPC-1-genet. Proteinet MRF er en såkalt transkripsjonsfaktor. Ved genkoding spiller det en rolle i dannelsen og beskyttelsen av myelinskjeder.
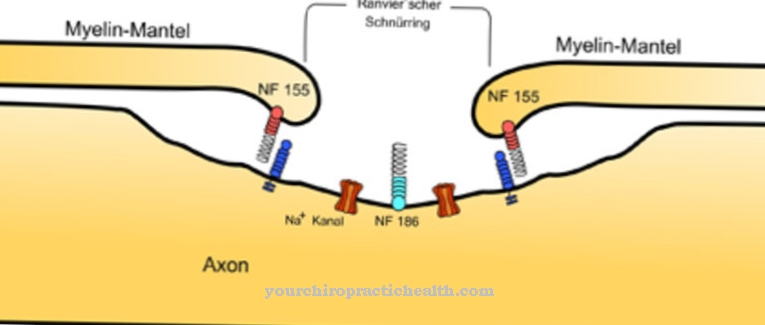
Myelin-skjeder dekker nervefibrene og sikrer at stimuli overføres raskt. Antagelig er de nevrologiske manglene som oppstår ved Niemann-Picks sykdom, basert på en feilaktig differensiering av oligodendrocytter. Disse cellene tilhører gliacellene. Deres celleprosesser dekker celleprosessene til nervefibrene som myelin-skjeder. Dermed fører den mangelfulle differensieringen av oligodendrocyttene til mangel på eller utilstrekkelig myelinisering.
I tilfelle av Niemann-Pick type C sykdom er kolesterolmetabolismen også nedsatt. I tillegg til sfingomyeliner, akkumuleres også kolesterol og andre metabolske produkter her i kroppens celler.
Symptomer, plager og tegn
Niemann-Picks sykdom kan deles inn i tre former:
- Type IA er også kjent som den akutte infantile nevropatiske formen. Sykdommen begynner i en alder av tre måneder og manifesterer seg som en drikkesvakhet og utviklingsforstyrrelser i individuelle vev og organer.
Det viktigste symptomet er hevelse i leveren (hepatomegali). Dette kan også oppstå i kombinasjon med en hevelse i milten (milteneglering). I tillegg kan lymfeknuter merkes og brun misfarging av huden skjer. Nevrologisk nedbrytning begynner i det andre året av livet. Berørte små barn blir døve, blinde og mister sosial kontakt.
Prognosen er dårlig, noe som betyr at alle barn med Niemann-Pick type IA sykdom vil dø i løpet av to år. Denne formen er den vanligste varianten av sykdommen.
- TYPE IS er også kjent som den kroniske viscerale formen. Det er et mildt forløp med hevelse i lungene og lungeinfiltrater. Det er ingen involvering av sentralnervesystemet. Forventet levealder for pasienter er bare litt begrenset.
- I type C av Niemann-Picks sykdom forekommer nyfødt gulsott. Huden og sklera hos de berørte nyfødte farges gule av avleiringer av fargestoffet bilirubin. Supranukleær parese er også typisk for denne varianten av sykdommen. Dette fører til progressiv lammelse av øyemuskulaturen med dobbeltsyn eller balanseforstyrrelser.
Cerebellar ataksi med nedsatt bevegelseskoordinasjon kan også observeres. I løpet av sykdommen utvikler pasienter ofte svelgforstyrrelser. Dette kan forårsake aspirasjons lungebetennelse. Utbruddet av sykdommen i type C er veldig varierende. De første symptomene kan vises hos spedbarn, barn eller til og med i ungdom eller voksen alder.
Diagnose og sykdomsforløp
Hvis risikoen for sykdommen er kjent, er prenatal diagnose mulig. Hvis det er mistanke om Niemann-Picks sykdom, tas hvite blodlegemer fra benmargen. Disse virker vakuolerte. Dette betyr at leukocyttene har hulrom. Det er også vakuolerte skumceller.
Dette fenomenet er kjent som "havblå histiocytose". Mangelen på aktivitet av enzymet sfingomyelinase kan påvises i kulturene til leukocytter og fibroblaster. Hvert andre barn med Niemann-Picks sykdom viser et rødt makulært merke under en okulær fundus.
komplikasjoner
Avhengig av type, er Niemann-Pick sykdom assosiert med en rekke komplikasjoner. Med TYPE IS kan leversvelling og lungeinfiltrater, dvs. akkumulering av fremmedlegemer i lungene, forekomme. Forventet levealder for de berørte er litt begrenset, og livskvaliteten er noen ganger sterkt nedsatt. Med type C kan de første symptomene vises i spedbarnsalderen.
Dette kan føre til alvorlige utviklingsforstyrrelser, som ofte er assosiert med cerebellar ataksi med forstyrrelser i bevegelseskoordinasjon. I løpet av sykdommen forekommer noen ganger svelgforstyrrelser, som fører til aspirasjons lungebetennelse og andre komplikasjoner. De berørte viser noen ganger symptomer på kortpustethet, som er assosiert med hoste med sputum, økt kroppstemperatur og en blå misfarging av hud og slimhinner.
I sin tur er slik cyanose fylt med alvorlige komplikasjoner. I TYPE IA er det tidlig drikkesvakhet og utviklingsforstyrrelser i organer og vev. Hevelsen i leveren er vanligvis assosiert med en hevelse i milten, noe som forårsaker alvorlig fysisk svekkelse hos de berørte.
Infeksjoner forekommer oftere, mage-tarmkanalen blir betent, og kroppens egne funksjoner avtar raskt. De berørte små barna blir vanligvis døve og blinde i løpet av to år før de til slutt dør av de alvorlige komplikasjonene av Niemann-Pick sykdom.
Når bør du gå til legen?
Niemann-Pick sykdom er en arvelig sykdom som tar et progressivt forløp. Foreldre som opplever at barnet deres har tilbakevendende gulsott og ubehag i muskler, bør kontakte barnelege. Hvis det er forsinkelser i motorisk utvikling eller psykologiske atferdsforstyrrelser, er mistanken om en alvorlig sykdom som må diagnostiseres og behandles åpenbar.
Foreldre eller foresatte bør besøke et spesialisert senter for sjeldne metabolske sykdommer. Barn med Niemann-Pick syndrom trenger pågående medisinsk behandling på grunn av økende fysiske og mentale problemer.
Uvanlige symptomer eller en plutselig økning i typiske symptomer må rapporteres til ansvarlig lege. Det samme gjelder hvis barnet ikke lenger tåler den foreskrevne medisinen eller viser andre avvik fra normal oppførsel. Rutinemessige behandlinger som stopper medisiner og fysiske undersøkelser kan utføres av fastlegen din.
De fleste mennesker med Niemann-Pick sykdom må behandles av spesialister på metabolske sykdommer. De enkelte symptomene blir behandlet av nevrologer, ortopeder og logopeder. I tillegg er fysioterapeuter og ergoterapeuter involvert i behandlingen. En terapeut kan også bli kalt inn for psykologiske klager som depresjon eller vrangforestillinger. På grunn av det store antallet mulige symptomer, må Niemann-Pick sykdom vanligvis behandles av et team av leger.
Terapi og behandling
En årsaksterapi er foreløpig ikke kjent. Imidlertid er det bevis for at spesielle syklodextriner kan lindre symptomene på sykdommen. Syklodextriner er sykliske oligosakkarider som ofte brukes som løsemidler i medikamentproduksjon. Niemann-Pick type C-sykdom behandles med miglustat.
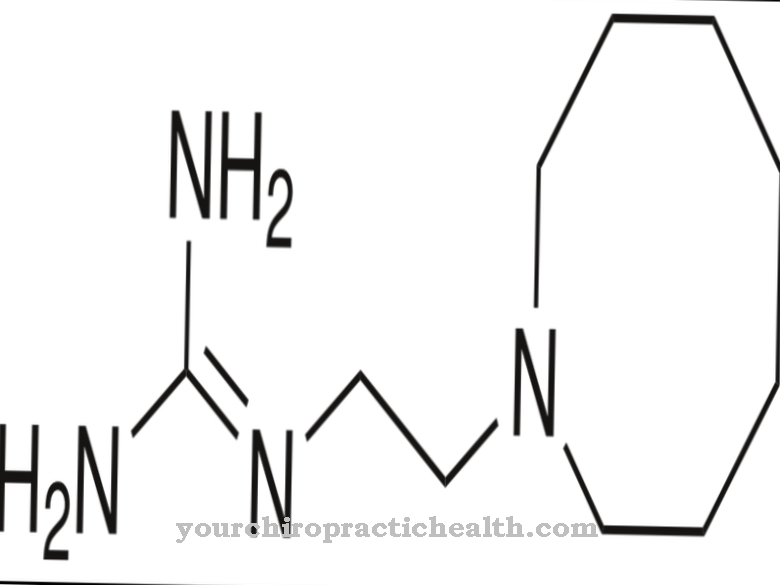
Miglustat er et medikament som bare er godkjent i EU for behandling av Niemann-Pick sykdom og for behandling av Gauchers sykdom type 1. Legemidlet er et iminosugar og et n-butylderivat av moranolin.
Du finner medisinene dine her
➔ Medisiner mot smerterOutlook og prognose
Prognosen for Niemann-Pick sykdom er dårlig. Sykdommen er en genetisk defekt. Gjeldende lovgivning forbyr forskere å blande seg inn i eller endre menneskelig genetikk. Selv om sykdommen kan diagnostiseres før fødsel, er ingen kur mulig på grunnlag av lovkrav.
Frem til i dag har leger og medisinsk fagfolk konsentrert seg om å utvikle tilstrekkelig medisinsk behandling etter at personen er født. Behandlingen består for tiden av å sette i gang medikamentell terapi for å støtte pasientens metabolisme best mulig. Som et resultat er optimeringer allerede mulig i pasientens utviklingsprosess, noe som bidrar til å forbedre den generelle situasjonen.
Uten behandling reduseres den berørte personens livskvalitet kraftig. I tillegg kan livstruende tilstander utvikle seg, da sykdommen er ledsaget av hevelse i indre organer og kortpustethet. Risikoen for en akuttsituasjon økes betydelig uten behandling. Langtidsbehandling indikeres derfor uavhengig av intensiteten til de enkelte symptomene. Pasientene trenger daglig pleie og støtte i å takle hverdagen. Avhengig av hvilken type sykdom som er til stede, kan sykdommen utvikle seg for tidlig i løpet av de første leveår hvis sykdommen utvikler seg dårlig.
forebygging
Niemann-Picks sykdom er arvelig som en autosomal resessiv egenskap. Det er foreløpig ingen effektiv forebygging.
ettervern
I de fleste tilfeller har den berørte bare noen få og bare begrensede oppfølgingstiltak tilgjengelig for Niemann-Pick sykdom. Av denne grunn må pasienten oppsøke lege ved de første tegn og symptomer, slik at det ikke er andre komplikasjoner eller klager. Jo tidligere en lege blir kontaktet, jo bedre er sykdomsforløpet, slik at en lege bør konsulteres så snart de første symptomene eller tegnene vises.
Hvis pasienten ønsker å få barn, bør genetisk testing og rådgivning definitivt gjennomføres for å forhindre gjentakelse av Niemann-Pick sykdom. De fleste av pasientene er vanligvis avhengig av inntak av forskjellige medisiner.
Personen som rammes, bør alltid ta hensyn til riktig dosering og også til regelmessig inntak for å lindre symptomene permanent. Hvis noe er uklart, eller hvis du har spørsmål, må du først kontakte lege. På samme måte er mange av pasientene avhengige av hjelp og støtte fra sine egne familier i hverdagen. Fremfor alt kan depresjon og andre psykologiske plager lindres.
Du kan gjøre det selv
Mulighetene for selvhjelp er ekstremt begrenset med Niemann-Pick sykdom. Spesielt type IA gir ikke tilstrekkelige muligheter til å forbedre situasjonen. Forventet levealder for det syke barnet er til tross for all innsats.
I hverdagen skal fokuset derfor være på å gjøre tiden sammen så hyggelig som mulig. Å glede seg over fritid er viktig for å bygge nærhet, solidaritet og stabilitet. Sykdommen er en enorm utfordring for både pasienter og pårørende. Å bygge opp mentale krefter er spesielt viktig når du takler motgang. Av denne grunn er psykologisk støtte viktig for alle involverte.
For mange er det også en hjelp hvis det er mulighet for utveksling med andre berørte personer. Det kan derfor være en fordel å kontakte etablerte selvhjelpsgrupper. I felles diskusjoner finner en utveksling sted på grunnlag av gjensidig forståelse. Kommunikasjon kan hjelpe med prosessering. Det gir også tips for å takle godt.
Psykiske teknikker og avslapningsøvelser fremmer reduksjon av stressorer. Siden situasjoner med overdreven stress og dermed vegetative problemer ofte oppstår, kan treningsenhetene bidra til å redusere stress. Håndteringen av den generelle situasjonen bør derved forbedres.



.jpg)

.jpg)

.jpg)




.jpg)



.jpg)