Som homocystinuria er en sjelden, genetisk metabolsk sykdom som skyldes en enzymmangel og er preget av en økt homocysteinkonsentrasjon i blodet. Homocystinuria kan vanligvis behandles godt med tidlig og konsekvent terapi.
Hva er homocystinuria?

© Anastasia Okhrimenko - stock.adobe.com
Som homocystinuria er en sjelden, genetisk bestemt aminosyremetabolismesykdom, som skyldes feil i forskjellige enzymer involvert i metioninmetabolisme (essensiell aminosyre).
Homocystein og homocystine er nedbrytning eller mellomprodukter av denne metabolske prosessen og metaboliseres umiddelbart videre hos friske mennesker. På grunn av tilstedeværelsen av mangelfulle enzymer er dette bare mulig i begrenset grad hos de som er berørt av homocystinuri, slik at konsentrasjonen av homocystein i blodet og homocystin i urinen økes.
Den økte konsentrasjonen av disse aminosyrene, som anses som giftige, kan skade forskjellige organsystemer. Øyesykdommer (linsedislokasjon, nærsynthet, glaukom), skjelettforandringer (osteoporose, marfanoide lange lemmer), skade på sentralnervesystemet (mental og fysisk utviklingshemning, spasmer, cerebrovaskulære lidelser) og det vaskulære systemet (tromboembolisme, vaskulær okklusjon) er karakteristiske følgene av homocystinuria.
fører til
homocystinuria skyldes en autosomal recessiv arvelig genetisk defekt, noe som resulterer i mangel på forskjellige enzymer involvert i metioninmetabolisme. Avhengig av det spesifikt berørte enzymet og underprosessen med metioninmetabolisme, skilles tre former for homocystinuri.
Med de hyppigere Type I. Ved homocystinuria er det en mangel i enzymet cystathione beta synthase (CBS), som forstyrrer syntesen av cystein fra metionin. Som et resultat bygger homocystein seg opp i blodet (hyperhomocysteinemia) og homocystine bygger seg opp i urinen (homocystinuria).
Type II Homocystinuria er preget av mangel på 5,10-metylen-tetrahydrofolatreduktase (MTHFR), som regulerer metioninsyntese fra homocystein. Denne metabolske underprosessen forstyrres tilsvarende hos pasienter av type II, og i tillegg til berikelse av serumet med homocystein, kan det også føre til en metioninmangel.
Type III homocystinuria er preget av en kobalaminmangel (koenzym vitamin B12). Cobalamin er også involvert i metioninsyntesen fra homocystein, slik at en mangel også kan forårsake en økt homocystein-konsentrasjon i blodet og en metioninmangel.
Symptomer, plager og tegn
Homocystinuria kan forekomme i forskjellige former. Symptomene er varierte og varierer avhengig av livsfase. Før fylte to år vises tegn på sykdommen bare i spesielt sjeldne tilfeller. Bortsett fra karakteristiske laboratoriefunn er nyfødte med homocystinuri normalt.
Typisk er homocysteinnivået i blodet betydelig økt. Dette skader blodkarene, som på lang sikt kan føre til vaskulær forkalkning (arteriosklerose) og den tilhørende emboli og trombose. Som et resultat er forventet levealder for de berørte betydelig. Det mest merkbare symptomet på metabolsk lidelse i barndommen er en prolaps av øyelinsen.
Dette er ofte assosiert med nærsynthet. Jo tidligere de første tegnene på sykdommen dukker opp, jo høyere er risikoen for psykomotorisk utviklingshemning (intellektuell funksjonshemning), som er irreversibel. De fleste av de berørte har allerede osteoporose i barndommen. Som et resultat flater ryggraden ut og deformeres gradvis.
Det høye homocysteinnivået fører ofte til høy status og symptomer som kan være lik Marfans syndrom. Disse inkluderer tilstedeværelsen av et kylling- og traktbryst, en fortrengt øyelins (linsedislokasjon eller linsedopati), glaukom (glaukom), retinal løsgjøring og edderkoppfingre (arachnodactyly).
Diagnose og kurs
Å diagnostisere homocystinuria forskjellige laboratorieanalyser brukes. Hvis en urinanalyse (f.eks. Cyanid-nitroprussid-test) avslører en økt homocystinkonsentrasjon og / eller en redusert metioninkonsentrasjon (type II og III), kan dette indikere homocystinuria.
En blodanalyse kan brukes til å bestemme serumhomocysteinkonsentrasjonen og diagnostisere hyperhomocysteinemia assosiert med homocystinuri. Diagnosen bekreftes ved dyrking av celler fra en bindevev eller levervevsprøve, hvorved den underliggende genetiske defekten kan oppdages direkte.
Forløpet av homocystinuria kan variere fra person til person når det gjelder symptomer og sekundære sykdommer. Med en tidlig diagnose og en tidlig start av terapi har homocystinuri vanligvis et gunstig forløp og en god prognose.
komplikasjoner
Homocystinuria fører først og fremst til alvorlige psykologiske plager som kan ha en ekstremt negativ effekt på pasientens liv og hverdag. I de fleste tilfeller oppstår en sterk personlighetsforstyrrelse, som er ledsaget av atferdsforstyrrelser. Disse lidelsene kan føre til alvorlige komplikasjoner, spesielt hos barn.
Som regel påvirkes pasienten av sosial eksklusjon og trekker seg mer og mer ut av livet. Det er ikke uvanlig at dette fører til depressive stemninger. Videre er det ubehag i øynene, slik at for eksempel en glaukom eller nærsynthet kan utvikle seg. Ulike sykdommer i blodårene forekommer også mye tidligere og kan føre til begrensninger i bevegelse.
Behandlingen i seg selv fører som regel ikke til spesielle komplikasjoner og utføres ved hjelp av medisiner. Sykdommen utvikler seg relativt raskt. I de fleste tilfeller er det ingen ytterligere symptomer, selv etter behandlingen. Tidlig behandling reduserer ikke forventet levealder. Psykologiske klager kan behandles støttende av en psykolog.
Når bør du gå til legen?
Hvis det oppstår symptomer som atferdsforstyrrelser, trombose eller forsinkelser i utviklingen, bør en lege absolutt sees. Tegn på osteoporose eller arteriosklerose bør også avklares på et tidlig tidspunkt. En lege må bestemme årsaken til symptomene og om nødvendig starte behandlingen. Derfor bør symptomene som er nevnt raskt avklares. Personer med en genetisk defekt er spesielt utsatt for å utvikle homocystinuria. De som rammes, bør konsultere huslege nøye og informere dem om uvanlige symptomer.
I utgangspunktet må klager avklares som vedvarer i mer enn noen få dager eller øker i intensitet over lengre tid. De karakteristiske tegnene på homocystinuri utvikler seg vanligvis lumske og blir ofte bare gjenkjent når irreversible sykdommer allerede har satt seg inn. Det er desto viktigere å gjenkjenne de tidlige symptomene og få dem behandlet. Personer som merker fysiske eller mentale forandringer i seg selv eller andre som kan være relatert til metabolske lidelser, bør snakke med familielegen så snart som mulig. Andre kontakter er spesialister i indremedisin eller spesialistklinikk for arvelige sykdommer.
Leger og terapeuter i ditt område
Behandling og terapi
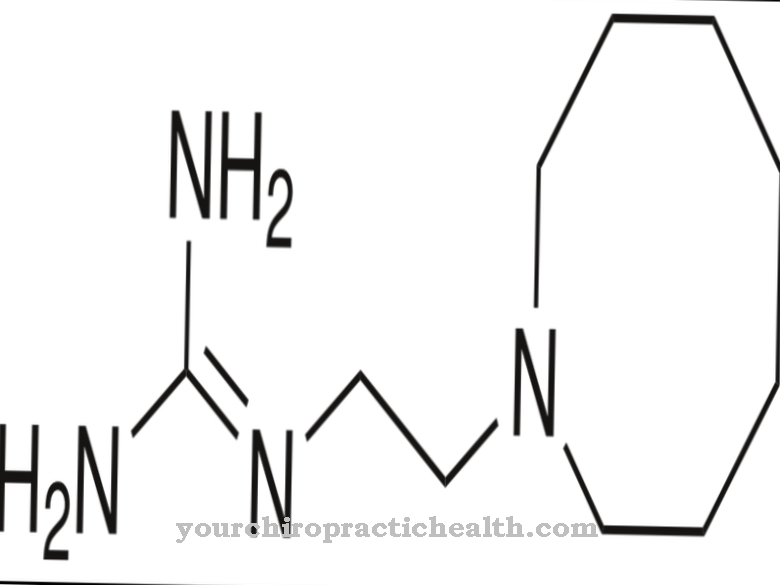
Terapien en homocystinuria avhenger av den underliggende sykdomstypen eller enzymdefekten og har som mål å redusere og eliminere den økte konsentrasjonen av giftig homocystein. Homocystinuri av type I behandles med pyridoksin (vitamin B6) når det defekte enzymet er gjenværende aktivitet.
Stoffet øker enzymaktiviteten og senker homocysteinkonsentrasjonen i blodet. Rundt 50 prosent av de som er berørt av denne typen svarer veldig godt på oral terapi med høydose vitamin B6. I tillegg anbefales et kosthold med lite metionin og høyt cystin som støtte terapi.
Hvis det er gjenværende enzymaktivitet i type II og III homocystinuria, der metioninsyntesen fra homocystein forstyrres, blir det forsøkt å begrense svekkelsen med kobalaminpreparater (vitamin B12). For begge former for homocystinuri indikeres et metioninrikt kosthold.
I tillegg brukes folsyre, som også har en positiv effekt på aktiviteten til den mangelfulle 5,10-metylentetrahydrofolatreduktasen, så vel som metionin og betain terapeutisk for type II. Antikoagulasjonsmedisiner (acetylsalisylsyre) brukes for å forhindre vaskulære sykdommer som trombose og emboli.
Outlook og prognose
Ved tidlig diagnose og intensiv terapi er prognosen for homocystinuri vanligvis gunstig. Sykdommen er ikke kurerbar fordi den er en genetisk defekt. Som en del av terapien er det imidlertid mulig å redusere konsentrasjonen av homocystein og metionin permanent, noe som betydelig reduserer sannsynligheten for utviklingsforstyrrelser og komplikasjoner.
Graden av alvorlighetsgrad av homocystinuria kan varieres. Det er former for sykdommen som i utgangspunktet er iøynefallende og ellers er milde. Imidlertid er det også en større risiko for å utvikle arteriosklerose, trombose, emboli, hjerteinfarkt og hjerneslag fra 20 til 30 år.
Imidlertid, hvis homocysteinkonsentrasjonen allerede er veldig høy i spedbarnet, er det stor risiko for fysiske og psykiske utviklingsforstyrrelser hos barnet uten intensiv behandling. Et psykisk handikap kan allerede bli tydelig i de to første leveårene. De berørte barna lider ofte av osteoporose. Opptil 70 prosent av ubehandlede barn utvikler øyeproblemer, noe som tydeligst kommer til uttrykk i en prolaps av øyelinsen. Andre konsekvenser for øynene er glaukom, ekstrem nærsynthet, retinal løsrivelse og blindhet. Hvis alvorlige former for sykdommen blir behandlet for sent eller ikke i det hele tatt, vil tromboser og embolismer utvikle seg hos 30 prosent av alle pasienter under 20 år.
forebygging
Der homocystinuria er en genetisk sykdom, det kan ikke forhindres. Imidlertid, hvis behandlingen startes tidlig, kan komplikasjonene av homocystinuri forhindres eller begrenses. I tillegg har de berørte muligheten til å få testet sitt ufødte barn for homocystinuri som en del av en prenatal diagnose (fostervannsanalyse). Søsken til de berørte anbefales også å sjekkes for homocystinuria.
ettervern
Avhengig av hvilken type enzymdefekt som er årsaken til den metabolske forstyrrelsen, er en rekke tiltak nyttige og nødvendige som en del av oppfølgingen av homocystinuri. Ved homohystinuri type I må pasienten følge et kosthold rikt på vitaminer i tillegg til medisinsk administrering av vitamin B6. Vitamin B6 øker aktiviteten til det defekte enzymet og fører følgelig til en lavere konsentrasjon av homocystein i blodet.
Kostholdet må følges permanent slik at denne effekten opprettholdes. Det samme gjelder forbruket av mat rik på cystin og lite metionin. Ved type II homocystinuria må kostholdet også videreføres for å oppnå langtidseffekter. I tillegg gjelder jevnlige oppfølgingskontroller under oppfølgingen. Legen må sjekke aktiviteten til de berørte enzymer og justere terapien om nødvendig.
Siden homocystinuria vanligvis ikke er en alvorlig sykdom, er medisinsk kontroll hver tredje til seks måned. Ved alvorlige lidelser, bør en spesialist konsulteres månedlig etter at selve behandlingen er fullført. I tillegg må man være oppmerksom på uvanlige symptomer, da den metabolske forstyrrelsen kan forårsake andre sykdommer på lang sikt som må behandles.
Du kan gjøre det selv
Avhengig av hvilken type enzymfeil som ligger til grunn for homocystinuri og terapien legen bruker, kan pasienten gjøre noen få ting selv for å lindre symptomene.
For det første er en diett rik på cystin viktig. Offeret skal hovedsakelig konsumere ris, nøtter, soyabønner og havreprodukter. Den aktive ingrediensen finnes også i vannmeloner, solsikkefrø og grønn te. Legen vil også foreskrive vitamin B12-tilskudd for å begrense svekkelsen. Personen som er berørt kan støtte disse tiltakene ved å lage en metionrik ernæringsplan sammen med en ernæringsfysiolog og implementere den konsekvent. Mat med høyt proteininnhold som egg eller kjøtt bør unngås. Mat med lite protein er tillatt, inkludert frukt, grønnsaker og pasta med lite protein, brød eller mel fra spesialistbutikker. Dette kostholdet bør også støttes med forskjellige B-vitaminer og folsyre.
Etter at sykdommen har løst seg, bør pasienten ha ytterligere kontroller. Homocystinuria er en livslang sykdom som krever regelmessig evaluering i et spesialisert behandlingssenter. Tett overvåking gjør det mulig å identifisere metabolske problemer tidlig og behandle før komplikasjoner oppstår.
.jpg)

.jpg)

.jpg)

.jpg)




.jpg)



.jpg)