Cystisk fibrose (cystisk fibrose) er en arvelig sykdom som er mest vanlig blant hvite mennesker. Et typisk tegn på cystisk fibrose er dannelsen av tykt slim i lungene og bukspyttkjertelen. Som et resultat er de indre organene alvorlig nedsatt.
Hva er cystisk fibrose?

Cystisk fibrose (cystisk fibrose) er en metabolsk sykdom som hovedsakelig skyldes en genetisk defekt. Det antas nå at rundt 4 millioner tyskere har minst genet som er ansvarlig for arv. Symptomene er komplekse og vanskelige å gjenkjenne, da de ofte forveksles med andre tilstander som astma eller bronkitt.
For eksempel lider de som rammes ofte av lungebetennelse og kortpustethet. Forstyrrelser som påvirker vekst og er undervektige er typiske symptomer på sykdommen. For noen få år siden var forventet levealder for pasienter maksimalt 5 år, men nå kan barn som har fått diagnosen cystisk fibrose nå et gjennomsnitt på 40 år.
fører til
Årsaken til Cystisk fibrose ligger i en genetisk defekt som blir gitt videre. For at sykdommen skal bryte ut, må imidlertid begge foreldrene bære det mangelfulle genet og gi det videre til barnet. Siden arv foregår på en autosomal resessiv og ikke-dominerende måte, kan et par foreldre som bare en av dem har genet, få et perfekt friskt barn. Hvis begge foreldrene er rammet, er sannsynligheten for at barnet utvikler sykdommen én av fire.
Siden det mangelfulle genet nå kan lokaliseres nøyaktig, er det mulig å avgjøre om barnet vil bli sykt eller ikke før barnet blir født. På grunn av feilen blir membranproteinet satt sammen feil, slik at sekresjonene også påvirkes og får en tyktflytende konsistens. Dette fører til at cellene tørker ut og spyttkjertlene, svettekjertlene og kjertlene i lungene blir tilstoppet av den viskøse væsken. Hvis det defekte genet er arvet, kan utbruddet av cystisk fibrose ikke lenger forhindres.
Symptomer, plager og tegn
Symptomene på cystisk fibrose varierer veldig fra person til person og kan påvirke flere organsystemer: Det meste av tiden påvirkes fordøyelseskanalen og luftveiene. Kronisk hoste med slimproduksjon, nedsatt fordøyelse og tilhørende undervekt er de vanligste tegnene på sykdommen. Den første indikasjonen kan allerede være veldig tøff avføring hos nyfødte, noen ganger er dette ledsaget av en tarmhindring.
Som et resultat kan alvorlig flatulens, fet diaré og oppkast med nærvær av galle oppstå. Små barn har en stor mage, men ekstremitetene virker ekstremt tynne. Barna lider ofte av magesmerter, får utilstrekkelig vekt og har dårlig toleranse for fet mat. I ung alder utvikler de som rammes ofte diabetes mellitus.
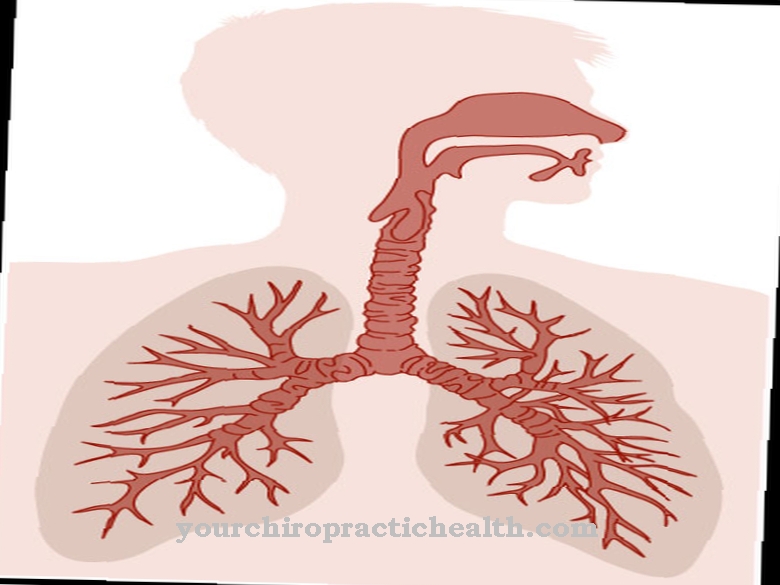
På grunn av den økte produksjonen av veldig tyktflytende slim i lungene, oppstår en nattlig irritabel hoste, som ligner en kikhoste og fører til alvorlig kortpustethet. Den skranglende pusten merkes, barna er ofte veldig rastløse. På grunn av den høye mottakeligheten for infeksjoner, kan den kroniske hoste gjentatte ganger bli til bronkitt eller lungebetennelse.
Noen ganger blødning fra lungene, som kan sees ved å hoste opp blodige slim. Neseslimhinnen har en tendens til å hovne opp, bihulebetennelser er ikke uvanlige og vanskeliggjør pusten. Nasale polypper er også mer vanlig hos barn med cystisk fibrose.
Forløp av sykdom
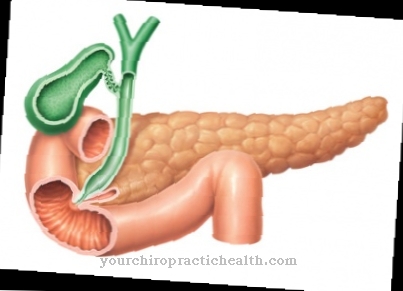
Er den Cystisk fibrose Når den har brutt ut, oppstår symptomer, som hovedsakelig skyldes det faktum at sekretene til den aktuelle personen blir tyknet og at det dannes en tyktflytende væske. Problemer oppstår derfor hovedsakelig i lungene og i bukspyttkjertelen.
Selv om sykdommen nå kan behandles på grunn av medisinsk fremskritt, er organenes funksjon fortsatt begrenset. Fremfor alt klager pasienter over ekstremt ubehagelig kortpustethet, som til og med kan føre til død ved kvelning. Symptomene blir vanskeligere og vanskeligere å tåle, da cystisk fibrose koster de som er rammet mye styrke over tid.
komplikasjoner
Ved cystisk fibrose lider de som rammes av hoste og alvorlig kortpustethet. Pustebesværet kan føre til svimmelhet eller panikkanfall og dermed redusere pasientens livskvalitet betydelig. Pustebesværet kan også føre til tap av bevissthet, noe som også kan skade personen det gjelder.
I tillegg opplever de berørte diaré eller flatulens og dermed betydelige begrensninger i hverdagen. Det er også smerter i magen. I verste fall kan pustevansker fortsatt føre til pasientens død. Som regel reduseres også pasientens spenst, og tretthet og utmattelse utvikles. Det er ikke uvanlig at de permanente smertene fører til depressive stemninger og andre psykologiske plager.
Dessverre er det ikke mulig å behandle cystisk fibrose årsakssammenheng. Imidlertid kan symptomene begrenses ved å spise riktig kosthold og ta medisiner. Det kan ikke forutsies universelt om cystisk fibrose vil føre til redusert forventet levealder. I mange tilfeller er de berørte også avhengige av avføringsmidler.
Når bør du gå til legen?
Et legebesøk er nødvendig hvis vedkommende lider av symptomer som hoste, alvorlig gass eller diaré. Hvis det gjentas utskillelse av slim når du hoster, bør du sjekke et lege hos en lege. Fordøyelsessykdommer, tap av matlyst og reduksjon i kroppsvekt er tegn på en eksisterende uregelmessighet som bør avklares av en lege. Hvis personen er sterkt undervektig, kan vedkommende utvikle en akutt helsetilstand uten tilstrekkelig medisinsk behandling.
Spesielt barn tilhører risikogruppen og er spesielt utsatt. Siden organismen er underforsynt med næringsstoffer, er det behov for handling og tilførsel i god tid i alvorlige tilfeller. Forstoppelse og tarmobstruksjon bør undersøkes og behandles. En kronisk hoste, tørr hoste og tap av normal ytelse er bekymringsfull.
Hvis vedkommende lider av en økt mottakelighet for infeksjon, diffus blødning eller pusteforstyrrelser, anbefales det et legebesøk. Det kreves lege i tilfelle pustelyder, vanskelig pust eller kortpustethet. Angst, humørsvingninger og atferdsproblemer bør også presenteres for en lege så snart de vedvarer over lengre tid eller øker intensiteten. Hvis det er generell ubehag, eller hvis de daglige kravene ikke lenger kan oppfylles som vanlig, trenger vedkommende medisinsk hjelp.
Behandling og terapi
Det kan sies at genetisk forskning ennå ikke har utviklet seg langt nok Cystisk fibrose ikke leges enda. Likevel er det måter å i det minste behandle symptomene og forlenge levetiden til de berørte. Det er mange forskjellige medisiner som kan bidra til å lindre noen av symptomene. Kosthold er imidlertid også viktig i behandlingen. Siden sykdommen tar mye styrke, bør mat som er høyt kalori (høyt kalori) spises, noe som gir pasienten nok energi.
Et annet problem er at enzymene fra proteiner og fett ikke absorberes ordentlig av kroppen, så disse gis best i form av kosttilskudd. Siden mange mennesker med cystisk fibrose ofte lider av fordøyelsesproblemer, kan laktulose, et avføringsmiddel, ha en positiv effekt på mage og tarm.
ettervern
Ved cystisk fibrose er det vanligvis ingen spesielle eller direkte tiltak og alternativer for oppfølgingspleie tilgjengelig for de som blir rammet, da dette er en arvelig sykdom. Av denne grunn bør pasienten oppsøke lege ved de første tegn og symptomer for å forhindre ytterligere komplikasjoner og klager.
Hvis du ønsker å få barn, bør genetisk testing og rådgivning også gjennomføres slik at sykdommen ikke kan komme igjen hos etterkommere og barn. De fleste med cystisk fibrose er avhengige av regelmessige kontroller og undersøkelser av en lege. Fremfor alt må de indre organene kontrolleres regelmessig for å identifisere og behandle skader på et tidlig tidspunkt.
Generelt bør den aktuelle personen ta hensyn til en sunn livsstil, der et sunt kosthold også er veldig viktig. Legen kan hjelpe med å lage en ernæringsplan. Forbruk av alkohol og sigaretter bør unngås. Medisiner kan også tas for tarmsykdommer, men vær alltid oppmerksom på riktig dosering. I de fleste tilfeller har personen som er rammet av cystisk fibrose ingen ytterligere oppfølgingstiltak tilgjengelig.
Du finner medisinene dine her
➔ Medisinering for kortpustethet og lungeproblemerOutlook og prognose
På grunn av det faktum at sykdommen er forårsaket av genetiske endringer, er den ikke kurerbar. Forventet levealder og livskvalitet blir vanligvis merkbart redusert. Uten spesiell terapi forverres helsetilstanden raskt, og de som rammes lever vanligvis ikke lenge uten behandling. Imidlertid er det mulig å redusere sykdomsforløpet betraktelig. Ved hjelp av konsistent og betimelig behandling lever nå pasienter betydelig lenger enn de gjorde for noen år siden. Gjennomsnittlig levealder er for tiden rundt 40 - 50 år. Imidlertid lever mange pasienter også med sykdommen mange år lenger.
Selv med intensiv terapi kan det imidlertid alltid oppstå noen komplikasjoner. Oftest lider de som rammes av akutt pustebesvær på grunn av dårlig lungeventilasjon.Individuelle deler av lungene kan også kollapse. Kronisk bronkitt eller lungebetennelse er vanlig. I tillegg kan sopp lett angripe lungene. I tillegg kan en forstyrret balanse i væske- og elektrolyttbalansen føre til sjokk og sirkulasjonssvikt.
Videre kan det i noen tilfeller forekomme redusert fruktbarhet hos kvinner og infertilitet hos menn. Pasienter bør søke genetisk råd hvis de ønsker å få barn. En genetisk test blir utført og det blir sjekket om CFTR-genet har noen endringer. Avhengig av dette kan det beregnes hvor høy risikoen for avkommet er.
Du kan gjøre det selv
Strengt overholdelse av behandlingsplanen som er foreskrevet av legen, er ekstremt viktig for å unngå komplikasjoner av cystisk fibrose og for å holde livskvaliteten stabil over lengre tid. Disse inkluderer inhalasjoner, regelmessige fysioterapiøvelser og individuelt tilpasset medikamentell terapi.
I tillegg spiller kosthold en viktig rolle: Et variert, kaloririk blandet kosthold anbefales, som kan berikes med sunt fett (f.eks. Vegetabilske oljer) på grunn av det økte energibehovet. I tillegg til de tre hovedmåltidene, bør pasienter med cystisk fibrose planlegge flere snacks mellom måltidene. I utgangspunktet er alt som smaker godt og stimulerer appetitten tillatt. Under måltider må pasienten ikke glemme å ta fordøyelsesenzymer slik at næringsstoffene kan tas opp av organismen.
Sportsaktiviteter kan også ha en positiv effekt på sykdomsforløpet. Utholdenhetsidretter som løping, sykling, fotturer, svømming og dans er spesielt godt egnet, og et besøk i treningsstudioet er også et alternativ. Før du begynner på trening, bør en individuell treningsplan utarbeides med den behandlende legen etter en grundig undersøkelse, som tar fysisk ytelse og spesielt lungekapasitet.
Det er også viktig for pasienter med cystisk fibrose å overholde strengere hygieniske regler for å redusere risikoen for lungebetennelse. Å vaske hendene grundig, skifte tannbørster, sengetøy og håndklær regelmessig og rengjøre inhalatoren forsiktig kan alt hjelpe.
.jpg)
.jpg)
.jpg)







.jpg)