De Kolesterol lagringssykdom er en lysosomal lagringssykdom og medfødt metabolsk sykdom med genetisk basis. Sykdommen er arvelig og er forårsaket av en genetisk mutasjon i genene som koder for lysosomal syrelipase. Den symptomatiske behandlingen av pasienten foregår konservativt med medisiner eller gjennom terapeutiske trinn for enzymerstatning.
Hva er kolesterolester lagringssykdom?

© fjerde livsfotografering - stock.adobe.com
Gruppen av lysosomale lagringssykdommer omfatter et antall medfødte sykdommer som kan føres tilbake til utilstrekkelig eller mangelfull aktivitet av de såkalte lysosomene. Alle sykdommer i gruppen er metabolske sykdommer. En slik sykdom er kolesterolesterlagringssykdommen, som ofte kalles CESD er forkortet.
Som med alle lysosomale lagringssykdommer, skyldes CESD også mangel på aktivitet av visse lysosomer. Mangelen på aktivitet i denne sykdommen knytter seg til lysosomal syrelipase, som bryter ned kolesterolestere og triacylglyserid hos friske mennesker. Kololerestinesterlagringssykdom er en ekstremt sjelden metabolsk sykdom og er assosiert med arvelighet. I denne sammenheng tilsvarer arv autosomal recessiv arv.
Sykdommen er en av de medfødte metabolske sykdommene. De første symptomene trenger ikke å manifestere seg umiddelbart etter fødselen. Den primære årsaken til sykdommen er en genetisk mutasjon. En sykdom med en lignende genmutasjon er Wolmans sykdom. I motsetning til denne sykdommen er kolesterolesterlagringssykdommen preget av et mye mildere forløp, ettersom en gjenværende aktivitet av lysosomalsyralipasen opprettholdes og kolesterolesternedbrytningen kan forårsake minst utenfor leveren.
fører til
Pasienter med kolesterolester-lagringssykdom lider av en enzymdefekt i lysosomal syrelipase. Denne defekten resulterer i en redusert nedbrytning av kolesterolestere. Dette resulterer i en ansamling av kolesterolestere og like lite nedbrytede triglyserider. Det kodende genet for sur lipase er lokalisert i DNA på kromosom 10 i genlokus q23.2 til 23.3. Ti eksoner utgjør genet.
Årsaken til koleretin esterlagringssykdom er en tull eller missense-mutasjon av de involverte eksonene. Rammeskift eller hopping av visse eksoner kan også være årsaken. Det muterte genproduktet viser redusert aktivitet, slik at knapt noen lipider i lysosomet kan trenge inn i cytoplasmaet. Kontrollløkken for regulering av intracellulære kolesterolkonsentrasjoner blir dermed avbrutt.
En intracellulær lavkolesterolkonsentrasjon oppstår og bringer den endogene kolesterolsyntese og LDL-reseptoraktiviteten til en oppregulering. Som et resultat tar lysosomet opp endocytosert kolesterol. På grunn av den endogene syntesen av kolesterol blir cellene overbelastet og lipidvakuoler dannes. Vakuolene fører til tap av funksjon av de enkelte celler, utløser fibrose og forårsaker celledød.
Du finner medisinene dine her
➔ Medisiner mot smerterSymptomer, plager og tegn
Kololerestinesterlagringssykdom er preget av et karakteristisk kompleks av kliniske symptomer. Lever abnormaliteter er spesielt typiske avvik. Som et organ er leveren involvert i metabolismen av kolesterol, noe som forklarer den foretrukne første manifestasjonen av sykdommen i leveren. Hos pasienter med kolesterolesterslagersykdom manifesterer seg symptomer mye senere i de fleste tilfeller enn hos pasienter med Wolmans sykdom.
I sistnevnte manifesterer de første symptomene seg mer eller mindre umiddelbart etter fødselen. I motsetning til dette kan pasienter med lagringssykdom for kolesterolestin forbli asymptomatiske i lang tid. Hovedsymptomet er hepatomegali. Leveren svulmer og utvikler seg til en fet lever i løpet av avsetningene. I tillegg forekommer hyperkolesterolemi i de fleste tilfeller.
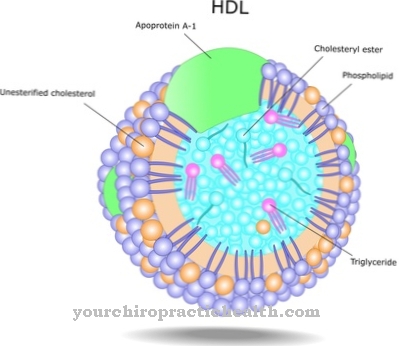
Denne hyperkolesterolemien tilsvarer en lipidmetabolismeforstyrrelse på grunn av økt kolesterolnivå i blodet. Disse symptomene er vanligvis assosiert med hyperlipidemia og en redusert HDL-konsentrasjon. I tillegg til hevelse i leveren og tap av funksjon derav, er de fleste av symptomene utelukkende diagnostiserbare ved laboratoriediagnostikk.
diagnose
For diagnostisering av kolesterolester-lagringssykdom er en laboratorieblodanalyse nødvendig, som kan oppdage det endrede lipidmønsteret og dokumentere skumceller som oppstår. I tillegg kan en leverbiopsi utføres som en del av diagnosen, som viser massive lysosmale ansamlinger.
Når det gjelder differensialdiagnose, må sykdommen differensieres fra andre, lysosomale lagringssykdommer innenfor rammen av diagnosen. Som regel blir dette skillet gjort i sammenheng med enzymatiske aktivitetstester. De genetiske testene for å identifisere mutasjonen brukes sjelden for differensiering.
komplikasjoner
Kolesterolesterlagringssykdom kan føre til forskjellige klager og komplikasjoner, som hovedsakelig avhenger av alvorlighetsgraden av sykdommen. I de fleste tilfeller påvirkes imidlertid leveren. Hos mange pasienter vises de første tegnene på kolesterolesterlagringssykdom umiddelbart etter fødselen, slik at for eksempel leveren er hovent eller hvis den senere utvikler seg til en såkalt fet lever.
Oftest opplever pasienter også smerter og svie fra hevelsen. Diagnostikk er vanligvis relativt enkelt med en blodprøve, så det er ingen forsinkelse i diagnosen. Dessverre er en kausal terapi og behandling av kolesterolesterlagringssykdommen ikke mulig, slik at fremfor alt symptomene må begrenses.
Dette resulterer i en redusert absorpsjon av kolesterol. Pasienten er imidlertid avhengig av bruk av medisiner hele livet. Ellers vil ikke kolesterolesterlagringssykdommen forårsake ytterligere klager eller komplikasjoner. Forventet levealder reduseres heller ikke med terapi. Pasientens hverdag er sjelden begrenset av sykdommen. Når du planlegger barn, bør imidlertid sannsynligheten for å tilbe sykdommen undersøkes.
Når bør du gå til legen?
Kolesterolesterlagringssykdom diagnostiseres vanligvis rett etter fødselen. Typiske tegn som indikerer sykdommen og som må avklares og behandles er en hovent lever og tegn på fet lever etter hvert som sykdommen utvikler seg. Hvis barnet klager på skarpe smerter i leveren, bør barnelege konsulteres umiddelbart. Dette gjelder spesielt hvis det er flere klager som indikerer kolesterolesterlagringssykdommen.
Ved alvorlige komplikasjoner, skal legevakt alltid kontaktes. Selv om livstruende symptomer sjelden forekommer med sykdommen, kan kronisk leversykdom utvikle seg. Derfor, ved de første tegnene på kolesterolesterlagringssykdom, bør du snakke med en lege.
Hvis det allerede er kjente tilfeller av sykdommen i familien, anbefales en undersøkelse rett etter fødselen. Under visse omstendigheter kan sykdommen også diagnostiseres prenatal. Legen vil deretter diskutere med foreldrene hvilke ytterligere tiltak som er mulig som en del av en behandling.
Leger og terapeuter i ditt område
Behandling og terapi
Kolerestinesterlagringssykdommen skyldes en genetisk defekt. Derfor er ingen hittil kausale terapeutiske trinn for behandling av pasientene. En kausal terapi ville bare være mulig i sammenheng med genterapimetoder. Genterapi har ennå ikke nådd den kliniske fasen. Derfor blir lagringssykdommen fortsatt ansett som uhelbredelig i dag og behandles bare symptomatisk. Symptomatisk terapi fokuserer på et redusert opptak av kolesterol.
For dette formålet blir pasientens tarmkolesterolabsorpsjon konservativt hemmet med medisiner. Legemidler som kolestyramin og ezetimibe er egnet for hemming. I tillegg får pasienter vanligvis statiner, som hemmer HMG-CoA-reduktase. I den siste tiden har det også blitt etablert nye behandlingsalternativer, spesielt enzymerstatningsterapier.
For pasienter med kolerestinesterlagringssykdom spiller enzymet sebelipase alfa en viktig rolle.Enzymerstatningsterapier med dette enzymet brukes for tiden i kliniske studier og ble godkjent og evaluert positivt av European Medicines Agency i fjor.
Outlook og prognose
Kolesterolesterlagringssykdom har en ugunstig prognose. Sykdommen anses som uhelbredelig og kan føre til vanskelige komplikasjoner. Den recessive arvelige sykdommen behandles symptomatisk av leger, fordi det av juridiske årsaker foreløpig ikke er tillatt å gjøre et inngrep i genetikk hos mennesker.
Alvorlighetsgraden av sykdommen er individuell og derfor forskjellig for hver pasient. Følgelig er det heller ingen enhetlig behandlingsplan. Når leveren er kompromittert, synker utsiktene til helseforbedring betydelig. En hevelse i leveren eller en fet lever kan føre til ytterligere sykdommer. Ofte er det økt betennelse eller levercirrhose. På dette stadiet er det nesten umulig å lindre symptomene. Den syke blir truet med leversvikt og dermed for tidlig død.
Pasienter hvis lever ikke har noen skade, har betydelig bedre helse. Så lenge de følger retningslinjene fra legene og unngår inntak av produkter som inneholder kolesterol i tillegg til medisiner, opplever de en god livskvalitet til tross for lagringssykdommen.
Siden behandlingen er en langvarig terapi, forverres helsetilstanden innen kort tid så snart legemidlet er avbrutt. En usunn livsstil har også en umiddelbar negativ innvirkning på pasientens velvære.
Du finner medisinene dine her
➔ Medisiner mot smerterforebygging
Kolerestinesterlagringssykdom er en genetisk sykdom. Siden ingen eksterne faktorer er kjent for å være årsaksfaktorer, kan sykdommen bare og bare forhindres gjennom genetisk rådgivning i familieplanleggingsfasen. Imidlertid kan dette heller ikke utelukke nye mutasjoner.
ettervern
I de fleste tilfeller er det ingen spesielle oppfølgingstiltak tilgjengelig for de som er rammet av kolesterolesterlagringssykdom. Pasienten er først og fremst avhengig av en rask diagnose slik at symptomene kan lindres på riktig måte, siden selvhelbredelse ikke kan forekomme. Derfor bør den berørte kontakte en lege så snart de første symptomene og klagene dukker opp for å forhindre at symptomene forverres.
Siden dette er en genetisk bestemt sykdom, bør det alltid foretas en genetisk undersøkelse og konsultasjon hvis du ønsker å få barn for å forhindre at kolesterolesterlagringssykdommen oppstår igjen. I de fleste tilfeller behandles sykdommen med medisiner.
Personen som rammes, bør sørge for at de tas regelmessig og at doseringen er riktig slik at symptomene blir permanent lindret. Kontakt lege først hvis noe er uklart eller hvis du har spørsmål. Med kolesterolesterlagringssykdom trenger mange pasienter hjelp og støtte fra venner og familie. Kontakt med andre mennesker som er rammet av sykdommen kan også være veldig nyttig.
Du kan gjøre det selv
Kolesterolesterlagringssykdom er en genetisk lidelse. Derfor er det foreløpig verken konvensjonelle eller alternative metoder for å behandle årsaken til sykdommen. Følgelig er det ingen tiltak for selvhjelp som kan bekjempe sykdommen årsakelig.
Imidlertid kan pasienter fremdeles gi et viktig bidrag for å unngå progresjon av sykdommen og alvorlige følgeskader, spesielt i leveren. Et lavkolesteroldiett er av sentral betydning. Pasienter bør innhente omfattende og kompetent informasjon om de spesielle diettkravene for kolesterolesterlagringssykdommen.
Den behandlende legen er ikke alltid den best mulige kontakten, siden ernæringsproblemer fremdeles neppe spiller noen rolle i opplæringen av leger. De berørte bør derfor oppsøke en ernæringsfysiolog eller en økotrofolog.
Generelt finnes kolesterol bare i matvarer av animalsk opprinnelse. Å bytte til et vegansk kosthold er derfor fornuftig for de berørte. Uansett bør mat av animalsk opprinnelse, som er spesielt høy i kolesterol, unngås.
Disse inkluderer spesielt fet kjøtt, pølseprodukter, innmat, egg, smør, fløte og helmelk. Egg blir ofte konsumert skjult. Pasientene er ofte uvitende om at mange typer pasta, spesielt pasta og kaker, samt ferdigretter og majones, inneholder store mengder egg og tilsvarende høye mengder kolesterol.
.jpg)
.jpg)
.jpg)






.jpg)