De Holt-Oram syndrom er et misdannelsessyndrom som primært er assosiert med hjertefeil og avvik i tommelen og er forårsaket av en mutasjon. I de fleste tilfeller forekommer den forårsakende mutasjonen sporadisk og tilsvarer dermed en ny mutasjon. Den kirurgiske korreksjonen av hjertedefekten er i fokus for terapien.
Holt-Oram syndrom?

© SmirkDingo - stock.adobe.com
De medfødte misdannelsessyndromene med overveiende involvering av ekstremitetene er en gruppe sykdommer som inkluderer forskjellige deformiteter i armer og ben. En av dem er dette Holt-Oram syndrom, også kalt Hjerte-hånd-syndrom er kjent. Syndromet er assosiert med gruppen av atriodigital dysplasi, som inkluderer medfødte sykdommer med misdannelser i de øvre ekstremiteter og hjertet.
I tillegg til Holt-Oram-syndrom, tilhører hjerte-hånd-syndrom type 2, hjerte-hånd-syndrom type 3 og hjerte-hånd-syndrom av den slovenske typen denne gruppen av sykdommer. Holt-Oram-syndromet ble først beskrevet i 1960. Den britiske barnelegen Holt og kardiologen Samuel Oram er de første som beskrev sykdommen. Holt-Oram-syndromet Dette er en relativt sjelden sykdom og er assosiert med en gjennomsnittlig utbredelse av en berørt person hos 100 000 mennesker.
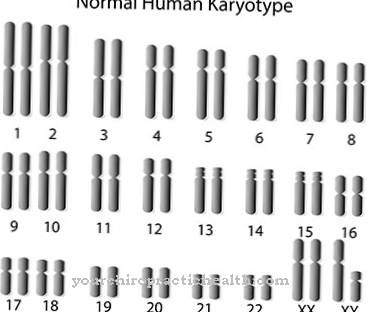
Årsaken til misdannelser i hendene og hjertet i Holt-Oram syndrom ligger i genetikk. Symptomene på syndromet er like varierte som det antas å forårsake. Misdannelsene i sykdommen konsentrerer seg om hjerte og tommel.
fører til
Selv om Holt-Oram syndrom er en genetisk og medfødt sykdom, er det knapt noen familiær hyppighet i de tilfeller som er dokumentert så langt. Selv om syndromet ser ut til å være arvet i en autosomal dominerende arvemodus i enkelttilfeller, antyder en stor del av saksdokumentasjonen dets sporadiske forekomst. Rundt 85 prosent av de dokumenterte tilfellene ser ut til å skyldes en ny mutasjon.
Den primære årsaken til Holt-Oram-syndromet ligger i genetiske mutasjoner ved genlokuset 12q23-24.1. Det såkalte TBX5-genet, som er lokalisert på kromosom 12 og er et protein involvert i lemmen og lemmen, er lokalisert på dette genlokuset. Kodet hjerteutvikling. De nøyaktige funksjonene til proteinet er ennå ikke avklart. Det er heller ikke ennå fastslått om eksterne faktorer som eksponering for giftstoffer eller underernæring hos mor under graviditet favoriserer mutasjonen av TBX5-genet.
Mutasjoner i genet kan påvises hos minst opptil 70 personer av 100 pasienter med Holt-Oram syndrom. Imidlertid antar vitenskapen at abnormiteter i andre gener også kan forårsake symptomene på syndromet. For eksempel er malformasjonssyndrom assosiert med treparts tommelpolysyndaktisk.
Symptomer, plager og tegn
Pasienter med Holt-Osram syndrom lider av et kompleks av misdannelser som først og fremst påvirker tommelen og hjertet. Selv om lokaliseringen av pasientens misdannelser er vanlig, er forskjellige typer misdannelser på hjerte og tommel mulig. Det kliniske bildet er derfor ekstremt variert.
Hjertedefektene kan manifestere seg, for eksempel i form av en ventrikulær septaldefekt, en atrisk septumdefekt, en hjertearytmi eller en ledningsforstyrrelse. Skjelettavvikene kan tilsvare reduserte misdannelser i tommelen, men avvik som ikke påføringen av eiken forekommer også.
Mange pasienter med syndromet lider også av radioulære synostoser og avvik i ribbeina, scapula eller clavicle. I tillegg er Holt-Oram syndrom assosiert med pectus carinatum og skoliose. Flertallet av de berørte lider også av syndaktylia i fingre eller tå faller. I enkelttilfeller er disse symptomene assosiert med hypertelorisme.
Diagnose og sykdomsforløp
Holt-Oram syndrom blir ofte feildiagnostisert. I en differensialdiagnose må legen differensiere symptomkomplekset fra Okihiro syndrom etter genmutasjoner i SALL4-genet på kromosom 20, som er assosiert med de samme arm misdannelser og hjertefeil. Det som er spesielt relevant ved den differensielle diagnosen er at pasienter med Okihiro syndrom vanligvis har en Duane-anomali.
De myser, er ofte rammet av nyremissdannelser og har hørselsforstyrrelser, fotanomalier eller øreproblemer. Trombocytopeni-fraværende-radius-syndrom må også differensieres fra Holt-Oram-syndrom, som hovedsakelig oppnås gjennom laboratoriediagnostikk. Andre kliniske bilder med et klinisk lignende bilde er Fanconi anemi og Pallister Hall syndrom.
Levealderen for pasienter med Holt-Oram syndrom er ikke under gjennomsnittet. Bare i alvorlige tilfeller er det en knapt behandlingsbar hjertefeil som gjør prognosen ugunstig.
komplikasjoner
Holt-Oram syndrom forårsaker en rekke forskjellige misdannelser og deformiteter hos pasienten, noe som kan gjøre livet og hverdagen vanskelig. Fremfor alt påvirkes hjertet av misdannelsene, slik at pasienten lider av en hjertefeil. Det er også en hjertearytmi som den rammede i verste fall kan dø fra.
Anomalier forekommer også på tommelen, slik at visse bevegelser eller prosesser i hverdagen også blir vanskeligere. Det er ikke uvanlig at deformiteter i kroppen fører til erting og mobbing av andre barn, noe som kan føre til psykologiske klager og depresjoner hos mange pasienter. Det er ikke uvanlig at nyresykdommer oppstår, som i verste fall kan føre til nedsatt nyrefunksjon.
Videre lider de berørte også av en synshemming og en hørselshemming. Som regel er forventet levealder uendret som et resultat av Holt-Oram syndrom, så lenge det ikke er noen hjertefeil som fører til død. En kausal behandling av Holt-Oram syndrom er vanligvis ikke mulig, så bare symptomene kan behandles. I mange tilfeller er psykologisk støtte også nødvendig.
Når bør du gå til legen?
Holt-Oram syndrom diagnostiseres vanligvis like etter fødselen. Avhengig av hvor alvorlige misdannelser er, kan det berørte barnet trenge ytterligere medisinske undersøkelser. I prinsippet må hjertedefekten behandles omgående for å redusere risikoen for alvorlige sekundære sykdommer. Hvis det utvikler seg komplikasjoner som hjertearytmier eller tegn på atrisk septumfeil, er medisinsk råd nødvendig. En medisinsk fagpersonell bør også konsulteres med avvik i skjelettens tommel.
Foreldrene til berørte barn bør konsultere legen sin nøye og informere dem om uvanlige symptomer. Siden Holt-Oram syndrom er en arvelig sykdom, er det ikke mulig å forårsake behandling. Pasientene kan derfor måtte behandles livet ut, avhengig av hvilke misdannelser som oppstår og hva pasientens konstitusjon er. Siden dette ofte også forårsaker emosjonelle klager, indikeres ledsagende psykologisk støtte. Barn som lider av mobbing eller erting bør søke terapeutisk rådgivning med foreldrene.
Leger og terapeuter i ditt område
Terapi og behandling
Ingen årsaksbehandlinger er tilgjengelige for pasienter med Holt-Oram syndrom. Det er håp om en kausal behandlingsevne i fremtiden, siden genterapi for tiden er gjenstand for medisinsk forskning. Så lenge denne typen terapi ikke når den kliniske fasen, er Holt-Oram syndrom imidlertid en uhelbredelig sykdom.
For øyeblikket er det bare symptomatiske behandlingsalternativer som er tilgjengelige for behandling av pasienter. Terapien er basert på symptomene i det enkelte tilfelle. Den tidlige korreksjonen av hjertedefekten er av spesiell relevans. Denne korreksjonen gjøres kirurgisk. Når det gjelder en Vohof septal defekt, tar den kirurgiske prosedyren sikte på å lukke den aktuelle feilen, for eksempel. Det samme er tilfelle for en ventrikulær septaldefekt.
Korrigeringer av misdannelser på ekstremitetene er i utgangspunktet av sekundær betydning. Etter vellykket korrigering av hjertefeilen, kan rekonstruktive kirurgiske prosedyrer gjenopprette manglende eiker og separate syndaktylier. En eksisterende skoliose bekjempes vanligvis under fysioterapeutisk pleie. I spesielt alvorlige tilfeller kan kirurgi for å implantere en titanribprotese være nødvendig.
I de fleste tilfeller er det ikke nødvendig med intervensjon for pectus carinatum. Av psykologiske årsaker kan imidlertid brystkassen omformes kirurgisk, for eksempel etter Nuss-prosedyren.
Outlook og prognose
Prognosen for Holt-Oram syndrom er gunstig. Selv om det er en genetisk defekt, kan den behandles tilstrekkelig med gjeldende medisinske alternativer. Levealderen til noen med syndromet er i de fleste tilfeller ikke under gjennomsnittet. Ved alvorlig misdannelse kan det være betydelige tap i forventet levealder. Prognosen er tydelig forverret hos disse pasientene. Hjertets aktivitet er begrenset og kan føre til en for tidlig levetid.
Imidlertid kan flertallet av pasientene behandles godt og vellykket. Selv om det ikke er noen kur på grunn av den genetiske defekten som er til stede, er det gode muligheter for forskjellige korreksjonsalternativer. Hjertets aktivitet er regulert og om mulig fullstendig korrigert i en kirurgisk prosedyre. Selv om det kan være en permanent svekkelse av levemåten i forhold til sunne mennesker, oppnås en god livskvalitet på grunn av behandlingen.
Fysiske avvik eller misdannelser endres i et ytterligere trinn. Etter at barnets vekstfase er fullført, initieres vanligvis en nødvendig eller ønsket korreksjon av de eksisterende misdannelsene. Hvis forstyrrelsene i utviklingsprosessen fører til betydelig svekkelse, blir det iverksatt tiltak i barndommen for å lindre symptomene. På grunn av de optiske endringene kan pasienten oppleve psykologiske konsekvenser. Dette forverrer den generelle prognosen.
forebygging
Så langt kan ikke Holt-Oram-syndromet forhindres fordi de ytre påvirkningsfaktorene ikke er blitt avklart.
ettervern
Siden Holt-Oram syndrom er en medfødt sykdom, kan det ikke kureres fullstendig. Derfor er tiltakene eller mulighetene for oppfølging er veldig begrenset, slik at den som rammes først og fremst er avhengig av tidlig oppdagelse og etterfølgende behandling. Hvis pasienten eller foreldrene ønsker å få barn, gis genetisk rådgivning for å forhindre at dette syndromet oppstår igjen.
Behandlingen av Holt-Oram syndrom er primært rettet mot å behandle hjertefeilen. Dette korrigeres ved en kirurgisk prosedyre, hvor pasienten må komme seg og hvile etter inngrepet. Anstrengelse eller fysisk aktivitet bør unngås. Det er ikke uvanlig at fysioterapeutisk behandling kreves, og mange av øvelsene kan også utføres i ditt eget hjem.
De berørte er noen ganger avhengig av hjelp og støtte fra sin egen familie og venner. Dette kan også forhindre psykologiske opprør eller depresjoner. Videre har en sunn livsstil med et sunt kosthold en veldig positiv effekt på forløpet av Holt-Oram syndrom.
Du kan gjøre det selv
Holt-Oram-syndromet kan ikke forhindres og kan heller ikke behandles med hjelp til selvhjelp. Med dette syndromet er de berørte alltid avhengige av en operasjon for å behandle hjertefeilen for å forlenge pasientens forventede levealder. Jo tidligere syndrom blir gjenkjent, jo større er sjansene for et positivt sykdomsforløp. De andre misdannelsene på kroppen må også korrigeres kirurgisk.
Siden mange av de berørte også lider av psykologiske klager eller av mindreverdighetskomplekser forbundet med dette syndromet, er de avhengige av psykologisk behandling. Imidlertid kan samtaler med andre pasienter, dine egne foreldre eller venner også styrke pasientens selvtillit og dermed lindre de psykologiske klagene. Videre er noen pasienter avhengige av hjelp fra sine medmennesker i hverdagen, hvor varm omsorg har en veldig positiv effekt på forløpet av Holt-Oram syndrom.
Siden sykdommen også kan påvirke de indre organene, er pasientene avhengige av regelmessige undersøkelser og kontroller fra forskjellige leger. Dette kan for eksempel forhindre nyreproblemer. Berørte barn skal informeres om konsekvensene og komplikasjonene av sykdommen.



.jpg)


.jpg)